





In vitro diagnostics (IVDs) are among the most commonly used medical devices in the world, and almost everyone has benefitted from an IVD at some point. From the strep tests you got as a kid with a sore throat to pregnancy tests and the blood glucose monitors that diabetics rely upon, IVDs are everywhere.
For IVD manufacturers as well as patients, the stakes related to these tests can be high. Millions of patients rely on IVDs for clarity, information, and an understanding of what’s next in their treatment. It’s essential that these tests perform the way they’re supposed to, every time.
In this guide, we’re going to walk through the basics of in vitro diagnostics, then dive deeper into the regulations that govern their design, development, and manufacture, as well as the unique characteristics that separate them from traditional medical devices.
Table of Contents
|
Are IVDs considered medical devices? |
|
How does FDA classify IVDs? IVDs and investigational device exemptions (IDE) |
|
How are IVDs regulated in the EU? How does the EU classify IVDs? Post-market requirements under IVDR |
|
Best practices for bringing an IVD to market QMS implementation |
|
Streamline your IVD journey with a MedTech-specific solution |
 What is an in vitro diagnostic (IVD) device?
What is an in vitro diagnostic (IVD) device?
In vitro diagnostics are clinical tests that analyze biological samples, such as blood, fluid, or tissue. IVDs are a common tool in healthcare, used in detecting disease or infection, measuring the concentration of specific analytes, or monitoring individual patient health.
In vitro means the tests are performed outside the body using test tubes, test kits, or related lab equipment. In fact, the latin term “in vitro” translates to “in glass”, denoting that IVDs use biological samples, rather than interacting directly with the patient. IVD samples can be collected and analyzed both in traditional healthcare settings, such as hospitals and testing labs, as well as remotely, such as at home or in the field.
Some common examples of in vitro diagnostic products include:
- Pregnancy tests
- COVID tests
- Blood glucose monitors
- Cancer diagnostics
- Devices for human genetic testing
- Devices for detection of infectious agents
- Immunoassays
After a spike in growth during the COVID-19 pandemic, the IVD market is projected to grow at a more steady 4.4% compound annual growth rate through 2030. Much of this growth can be attributed to rise in both chronic and infectious diseases and an increasing number of geriatric patients in key markets around the world.
Are IVDs considered medical devices?
The short answer is yes, IVDs are still considered to be medical devices and are regulated as such. There’s often some confusion around this, IVDs are given their own category (and often their own regulations) within the overarching medical device umbrella.
The US Food and Drug Administration (FDA) defines in vitro diagnostics as:
Those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.
The reason that IVDs have a separate regulatory scheme from other medical devices is due to the fact that they test “specimens taken from the human body,” rather than interacting directly with the patient.
This means that the intended uses for IVDs, and the types of risks they pose to patients, are typically different from traditional medical devices. A pacemaker, for instance, creates very different risks for a patient than that of a cancer diagnostic.
However, as we’ll see when we go through risk classifications for IVDs, that does not mean that IVDs are automatically low risk products. For many tests, the risk associated with a false positive or false negative is extremely high.
What is a companion diagnostic?
FDA defines an IVD companion diagnostic as “an in vitro diagnostic device that provides information that is essential for the safe and effective use of a corresponding therapeutic product.” For example, a companion diagnostic could be used in cancer treatment to determine whether a tumor has a specific gene change or biomarker that could be targeted by a specific drug.
Due to the essential nature of the diagnostic, FDA also states that the use of the companion diagnostic with the therapeutic product should be “stipulated in the instructions for use in the labeling of both the diagnostic device and the corresponding therapeutic product, including the labeling of any generic equivalents of the therapeutic product.”
Ideally, both the therapeutic product and the companion diagnostic should be developed contemporaneously, though FDA notes there are occasions when this isn’t possible.
As you might expect, companion diagnostics are high-risk by nature and typically require the most rigorous regulatory pathway.
Can software be an IVD?
Software can be an IVD and there are many companies out there developing software as a medical device (SaMD) IVDs. For example, a diagnostic tool that uses machine learning to inform the diagnosis of a mental illness based on patient responses would be considered a SaMD IVD.
In fact, a recent McKinsey report on the diagnostics industry projected the most growth in digital diagnostics, while the overall market for IVDs is projected to grow more slowly.
Software as a medical device is a huge topic. So much so that we’ve written a separate guide for SaMD that you should check out if you want to learn more. The key thing to remember, especially for those new to MedTech coming from the software realm, is that SaMD are legally classified as medical devices and you must follow all applicable regulations throughout the lifecycle of the device.
For companies that are heavy on software expertise, I would highly recommend having a medical device expert—someone who understands and can help navigate those regulatory requirements—on your team from the very beginning.
![in-vitro-diagnostic-ivd-devices-2]() How are IVDs regulated in the US?
How are IVDs regulated in the US?
 How are IVDs regulated in the US?
How are IVDs regulated in the US? As I mentioned before, IVDs are medical devices. And that means, in the US, they’re regulated in the same manner as traditional medical devices, meaning they are subject to both premarket and postmarket controls.
How does FDA classify IVDs?
As with traditional medical devices, FDA uses a three-tier classification system for IVDs.
- Class I devices pose low to moderate risk to the patient and/or user
- Class II devices pose moderate to high risk to the patient and/or user
- Class III devices pose a high risk to the patient and/or user
The big difference in risk classification with IVDs comes down to the way their risk is assessed. For traditional devices, like the pacemaker I mentioned earlier, risk to the patient comes from the device’s interaction with their body (the device fails to work properly, the device causes a biological reaction, etc.)
For IVDs, however, the risks associated with a given device are very different. An IVD doesn’t interact directly with a patient; it interacts with a specimen taken from the human body. This means that certain risks, like the devastating failure of a pacemaker to operate, are not an issue.
Instead, the risk involved with IVDs generally stems from the potential consequences of their inaccuracy. In other words, what is the overall risk associated with a false positive or a false negative? Obvious examples include cancer diagnostics or HIV tests, where the consequences of a false negative test can be life-threatening or pose a risk to public health.
But there are less obvious risks involved, as well. In an episode of the Global Medical Device Podcast, Christie Hughes, Principal Consultant and IVD Expert at Qserve Group, spoke about the consequences of a false positive test during newborn screening. Studies show that a false positive for a congenital disorder, even if it’s quickly corrected, can cause parents to treat that child differently for life. They may rush to the emergency room or visit clinicians more often due to lingering fear.
There is a cost, both financially and emotionally, to the results of that false positive that must be taken into account when determining the risk, and therefore the controls that must be in place, when developing an IVD.
How do you determine device classification for an IVD in the US?
The process for classifying an IVD in the US is the same as the process for classifying a traditional medical device. The classification for IVDs still stems from its intended use and indications for use:
-
Intended Use is the general purpose of the medical device or its function (what you “claim” the medical device does).
- Indications for Use describe the disease or condition the medical device will diagnose, treat, prevent, cure, or mitigate, including a description of the target patient population.
Remember, the intended use and indications for use of your medical device convey the reasons you developed this new medical device in the first place.
Once you define intended use and indications for use, your next step is to find the possible regulations and product codes via FDA databases. To find the regulation for your device, you can go directly to the FDA Product Classification Database and search for the device name. Or, if you know the speciality your device belongs to, you can go directly to the listing for that speciality and find your device and the corresponding regulation.
Finding the applicable regulation for your medical device and classification is the first part. Then you need to find the applicable product codes. To do so, go to the FDA Product Classification Database and type in the regulation number you found. If you find more than one possibility, then you will need to repeat this process for each.
When you click “search”, you will get a list of possible product codes. You can then review each individual code to determine the best option for your product by clicking on each code.
For a formal determination of device class, you can submit a 513(g) request to FDA. There is a fee, but if you’re genuinely unsure of your device class it may be worth it to get a determination directly from FDA.
What are the different pathways to market for an IVD in the US?
As with the classification process, the pathways to market for an IVD are the same as those for a traditional medical device.
510(k) premarket submission for IVDs
The 510(k) is a premarket submission in which a manufacturer demonstrates that their IVD is substantially equivalent to (and at least as safe and effective as) a device that is already legally marketed and not subject to premarket approval (PMA).
The review process will include an evaluation of the device’s analytical performance characteristics compared to the predicate device (the device already on the market), which includes:
- Bias or inaccuracy of the new device
- Imprecision of the new device
- The analytical specificity and sensitivity of the new device
In most cases, analytical studies using clinical samples are sufficient to demonstrate substantial equivalence. Prospective clinical studies for IVDs are rarely required in a 510(k), but are not out of the question.
Premarket approval (PMA) for IVDs
The PMA process is used to review the safety and effectiveness of Class III devices before they enter the market. Remember, for IVDs, a device’s safety is related to the impact of false negatives or positives on patient health.
The PMA process for IVDs operates in the same way as the process for traditional medical devices, and includes a PMA application and a lengthy scientific and regulatory review of the submission.
De Novo for IVDs
The De Novo process is for novel Class I or Class II devices that do not have a predicate device on the market. To be clear, any Class III device will need to go through the PMA process. But for IVDs without a predicate device on the market, manufacturers can submit a De Novo Classification Request to market their device.
Pre-Submission Process
The FDA’s Pre-Submission process allows IVD manufacturers to submit a formal written request for feedback from the agency around product development or application preparation.
FDA encourages manufacturers to use a Pre-Submission under certain circumstances, such as:
- The device involves new technology, a new intended use, or a new analyte and it will be helpful to familiarize the FDA with the novel features in advance of the submission;
- Assistance is needed in defining possible regulatory pathways;
- The studies involve complex data and/or statistical approaches;
- The predicate or reference method is unclear or uncertain; or
- The new device is a multiplex device capable of simultaneously testing a large number of analytes.
The benefit of the Pre-Submission process is that it allows a manufacturer to get the FDA’s thoughts on their studies or proposals, gives the agency a better understanding of the product, and ultimately facilitates the review process later on. The process is entirely voluntary, but it can be very useful for IVD manufacturers.
IVDs and investigational device exemptions (IDE)
In the US, an investigational device exemption (IDE) is required before a medical device that has not been cleared for marketing can be used in a clinical investigation. However, due to the nature of IVDs, many of them are exempt from the requirement to obtain an IDE before pursuing a clinical study. Note that you will still need IRB or EC approval before beginning your study.
IVDs are exempt from IDE if their testing:
- is noninvasive;
- does not require an invasive sampling procedure that presents significant risk;
- does not by design or intention introduce energy into a subject; and
- is not used as a diagnostic procedure without confirmation by another medically established diagnostic product or procedure.
However, even devices that are exempt from IDE requirements must comply with labeling requirements per 21 CFR Part 809.10(c) when under study.
What are LDTs and what are their current regulatory status?
Recently, FDA released its Final Rule on the phaseout of enforcement discretion for laboratory developed tests (LDTs). In other words, LDTs will be treated the same as other in vitro diagnostics, and regulated as such. Here is the stated timeline for the phaseout:
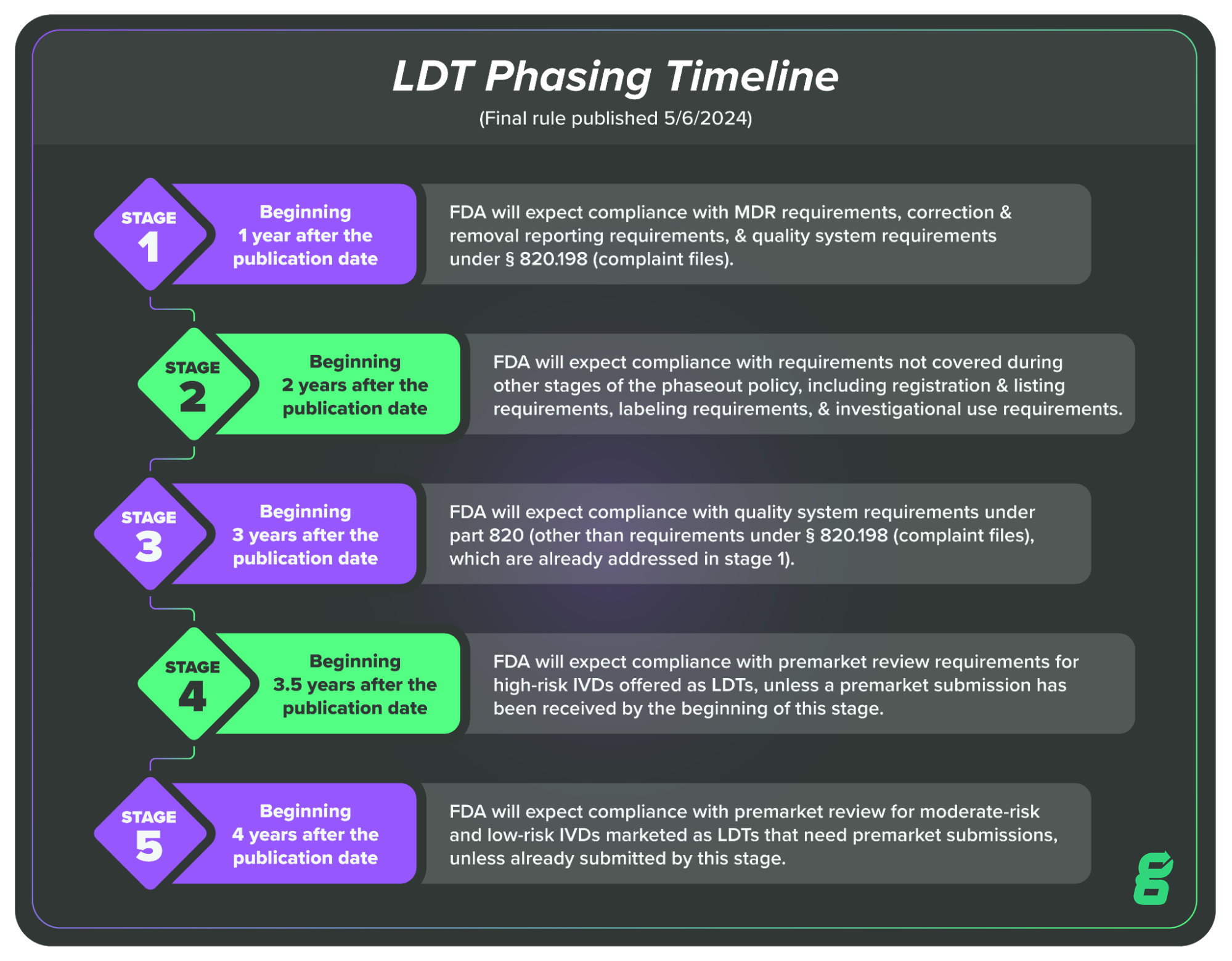
The bottom line is that over the course of the next few years, LDT manufacturers will need to begin complying with the regulations governing IVDs, including those surrounding quality systems.
There is some pushback on this rule currently happening, and it remains to be seen if changes will be made, but as of writing, it’s essential that LDT manufacturers begin preparing for these changes. And if you’re unfamiliar with the change, I’d encourage you to check out more of our resources on the topic.
FDA reclassification of high risk IVDs
Something else to be aware of: FDA recently began the process of reclassification for most Class III, high-risk IVDs, intending to downgrade them to Class II. The products being reclassified are mostly infectious disease and companion diagnostics, and FDA’s goal is to enable manufacturers of these devices to use the 510(k) pathway rather than using the more onerous PMA process.
FDA believes there is sufficient information available to establish special controls that ensure reasonable safety and effectiveness for these devices in addition to general controls. The stated reason for this change is to support more manufacturers in developing these tests which will increase competition and patient access.
But there is another element to the reclassification. Some suspect this is also related to the end of enforcement discretion applied to LDTs. In essence, FDA may be helping to reduce the number of LDTs that will need to go through the more rigorous PMA process when enforcement discretion finally ends.
The LDT saga is far from over, and we will update this if there are changes, but it’s worth keeping an eye on anything that affects risk class and market pathway.
![in-vitro-diagnostic-ivd-devices-3]() How are IVDs regulated in the EU?
How are IVDs regulated in the EU?
 How are IVDs regulated in the EU?
How are IVDs regulated in the EU?While it might seem like there’s plenty of regulation to go around in the US, there is even more to cover in Europe. That’s because back in 2017, the EU released the Medical Device Regulation (MDR) and the In Vitro Device Regulation (IVDR) to replace the old Medical Device Directives (MDD) and In Vitro Diagnostic Directives (IVDD).
IVDR has been applicable since May of 2022, however, as the European Commission noted in 2023, “Today a considerable number of in vitro diagnostics currently on the market do not yet comply with the new rules nor have been replaced by new devices.”
Therefore, the EC proposed to extend the transition period and the European Parliament agreed, voting in favor of the proposal in April 2024. The new timeline looks like this:
- High individual and public health risk devices such as HIV or hepatitis tests (class D) have a transition period until December 2027;
- High individual and/or moderate public health risk devices such as cancer tests (class C), have a transition period until December 2028;
- Lower risk devices (class B such as pregnancy tests and class A sterile devices such as blood collection tubes), have a transition period until December 2029.
The delays reflect the sheer number of requirements in the regulation and the potential difficulty that companies face in complying. However, we can’t pretend the regulation doesn’t exist, and it’s always a good idea to understand the fundamentals of how your product will be viewed and regulated in any market you’re considering.
How does the EU classify IVDs?
Similarly to the risk classification in the US, the EU system takes into account the risk to patients, as well as the risk to public health due to transmissible agents within biological material. The IVDR establishes four risk classes based on both patient and public health risk:
- Class A - Low patient and public health risk
- Class B - Moderate patient risk and/or low public health risk
- Class C - High patient risk and/or moderate public health risk
- Class D - High patient risk and high public health risk
Examples of IVD devices that fall into each of these four risk classification types include:
- Class A: Examples of Class A IVDs include specimen receptacles, laboratory instruments, and buffer solutions.
- Class B: Class B devices include IVDs for self-testing with less risk to the patient than those in Class C. For example, Class B devices include pregnancy tests, fertility tests, and cholesterol tests. Class B is also the default classification for IVDs that are not covered by any other rules.
- Class C: Class C devices include IVDs that are intended to be used for detecting an infectious agent without a high risk of propagation, or for detecting the presence of an infectious agent with the potential to cause death or severe disability in the case of an erroneous result.
- Class D: This device class includes IVDs that detect or are exposed to life-threatening transmissible agents or transmissible agents and infectious diseases with a high risk of propagation.
How to classify your IVD under IVDR
IVDR outlines seven rules for classification along with guidance on implementing the rules to help IVD manufacturers identify the risk class of IVD devices.
-
Rule One states that devices are Class D if they:
- Detect or are exposed to a transmissible agent in blood or tissue
- Detect or are exposed to a transmissible agent that causes a life-threatening disease with a high risk of propagation
- Determine the infectious load of a life-threatening disease in a situation where monitoring is of critical importance
-
Rule Two states that devices used for blood grouping or tissue typing to ensure the compatibility of blood, tissue, or organs for transfusion, transplantation, or cell administration are Class C, with some specific exceptions.
-
Rule Three lists the types of devices that are also Class C devices, including (but not limited to) those that:
- Detect or are exposed to a sexually transmitted agent
- Detect an infectious agent without a high risk of propagation
- Detect an infectious agent if there is a significant risk that an error would cause death or severe disability (For a full list of Class C sub-rules, see Annex VIII)
-
Rule Four states that devices for self-testing are classified as Class C, with several exceptions that include pregnancy tests, fertility tests and cholesterol tests. Those devices are classified as Class B.
-
Rule Five states that products for general laboratory use, instruments, and specimen receptacles are Class A.
-
Rule Six states that devices not covered by the previous rules are Class B.
-
Rule Seven states that devices which are controls without a quantitative or qualitative assigned value are Class B.
For the full text of the rules, see Annex VIII in the IVDR.
It should be noted that under the new rules, the majority of IVDs will be classified as Class B or Class C. However, IVDR also states that if a device has multiple intended purposes that fall under different classes, the device should be placed in the highest-risk class.
What are the different pathways to market for an IVD in the EU?
Under the old In Vitro Diagnostic Directives (IVDD), the vast majority of IVDs could be self-certified by their manufacturers, meaning they did not require the involvement of a Notified Body to obtain their CE Marking.
However, the requirements in IVDR mean that manufacturers now face the opposite situation—most of them will need to undergo a conformity assessment by a Notified Body. In fact, unless an IVD is a Class A, non-sterile device, it will now require the involvement of a Notified Body.
This has huge implications for IVD manufacturers. For one, it increases the number of companies that Notified Bodies must audit. In fact, the workload for Notified Bodies is one of the reasons for the delay in applying IVDR that we covered earlier.
Even with the delay, however, it’s crucial that IVD manufacturers understand there may be a bottleneck and should proactively work with a Notified Body during the extended transition period.
IVDR also requires every IVD manufacturer to implement a quality management system (QMS). Manufacturers of all device classes aside from Class A, non-sterile IVDs will need to have their QMS and their technical documentation reviewed by a Notified Body.
In the EU, ISO 13485:2016 is the recognized standard for a QMS, and an audit must be performed to make sure that your QMS conforms to the standard’s requirements. If the IVD manufacturer passes the audits, they will be issued an ISO 13485 certificate.
In order to obtain a CE marking (certification that the product has met the EU’s health, safety, and environmental requirements), the manufacturer must also undergo a review of their technical file, and any deficiencies must be resolved and closed.
Afterward, the manufacturer must formally document a declaration of conformity, as directed by Annex IV of IVDR. The Declaration of Conformity is the manufacturer’s assent that their device is compliant with all applicable regulations within the EU. At this point, the CE Marking may be affixed to the product.
Remember, all IVD device classes other than Class A, non-sterile will continue to have annual assessments of their QMS and technical documentation by their Notified Body to ensure their compliance with IVDR classification and other applicable requirements.
What are the post-market requirements for IVDs under EU IVDR?
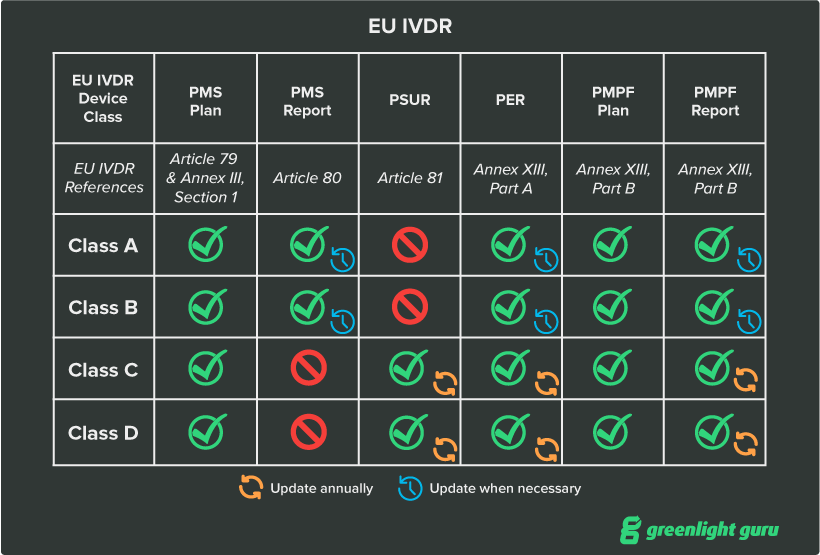
Post-market surveillance (PMS) takes a high priority within IVDR and depending on the risk class of your device, you’ll have different responsibilities with regards to PMS, with the objective of collecting data related to the quality, performance, and safety of the device to make informed decisions on overall improvement of the product. I’ve listed the major elements below:
- Post-market surveillance plan. The PMS plan is part of your device’s required technical documentation and details your strategy for continuously monitoring and collecting data and safety information on the device. The PMS plan is required for all IVD classes
- Post-market surveillance report (PMSR). The PMSR is a summary of the results and conclusions drawn from the data you generated as part of your PMS plan. As part of a device’s technical documentation, it primarily serves as proof of your compliance with MDR’s requirements for postmarket surveillance. The PMSR is required for Class A and B devices.
- Periodic safety update report (PSUR). The periodic safety update report is a summary of the results of postmarket surveillance activities as well as the conclusions that manufacturers have drawn from those results. PSURs are identical to PMSRs with a few additions—manufacturers must publish the conclusion of the benefit-risk determination, main findings of the post-market performance follow-up (PMPF), sales volume, and estimated user population characteristics and usage frequency.
- Performance evaluation report (PER). The PER is an assessment of the scientific validity, analytical performance, and clinical performance of your IVD based upon an associated performance evaluation plan. It must be updated at least annually for Class C and D devices and on a regular (and justified) schedule for Class A and B devices.
- Post-market performance follow-up (PMPF). The goal of the PMPF is to proactively collect and analyze performance evaluation data after the device is placed on the market. For Class C and D devices, the PMPF will most likely be required. It may not be required for Class A and B devices, but you must be able to justify the decision not to conduct a PMPF.
What does performance evaluation entail under IVDR?
IVDR defines Performance Evaluation as “an assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device.” The regulation goes on to state that PE is a continuous process that must be carried out throughout the life cycle of the device.
Scientific validity, analytical performance, and clinical performance are the three pillars of your PE and, in turn, you’ll need to prove each of them.
- Scientific validity - IVDR defines scientific validity of an analyte as “the association of an analyte with a clinical condition or physiological state.” For example, hemoglobin is correlated with anemia and other blood disorders. In this component of the PE, you will be expected to justify the use of any analytes, markers, or targets you use and establish their scientific validity via literature, clinical performance studies, or proof of concept studies.
- Analytical performance - Analytical performance refers to how well your IVD measures or detects your analyte. To evaluate the analytical performance of your IVD, you’ll need to carry out bench studies like cross-reactivity, interference, and stability testing. The full list of characteristics you’ll need to measure can be found in Annex I, Section 9.1 of IVDR.
- Clinical performance - IVDR defines clinical performance as “the ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user.”
In other words, clinical performance is a measure of your device’s diagnostic accuracy. You’ll likely need to conduct a clinical performance study to prove this, though you may use literature as supporting evidence.
The results of all three of these items must be documented in individual reports. IVDR addresses scientific validity, analytical performance, and clinical performance in a comprehensive manner in Annex XIII, and I’d strongly encourage you to read through it carefully.
MDCG guidance documents for IVDs
To help companies with the various complexities of MDR and IVDR, the European Commission’s Medical Device Coordination Group (MDCG) has been releasing guidance documents on various subjects related to the regulations. You can find the full list here.
Many of the guidances cover IVD topics, and some are quite helpful for MedTech companies trying to comply with IVDR. Here are some of the most useful:
-
MDCG 2022-2, Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs)
-
MDCG 2020-16 Rev.3, Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746
-
MDCG 2022-19, Performance study application/notification documents under Regulation (EU) 2017/746
-
MDCG 2022-3, Verification of manufactured class D IVDs by notified bodies
![in-vitro-diagnostic-ivd-devices-4]() Best practices for bringing an IVD to market
Best practices for bringing an IVD to market
 Best practices for bringing an IVD to market
Best practices for bringing an IVD to marketDepending on the device and its risk class, bringing an IVD to market can be a significant undertaking. Decisions around the regulatory pathway, necessary testing, and your quality management system can all affect your timeline. Here, I want to touch on some of the most important aspects of the device that you’ll need to consider.
Implement a QMS early on
Because in vitro diagnostics are medical devices, they need to follow many of the same regulations as traditional medical devices. This includes regulations such as the FDA’s Quality System Regulation (QSR), which can be found in 21 CFR Part 820.
This regulation, along with EU IVDR, require MedTech companies producing IVDs to implement a quality management solution (QMS) in order to ensure that their products are as safe and effective as possible. If you’re outside the US, the globally recognized harmonized standard, EN ISO 13485:2016 is the standard you’ll use for implementing your QMS.
If this is your first product, keep in mind that you don’t need an extensive QMS set-up on day one. You can look at your short-term goal to determine what parts of the QMS you need to focus on building first. Usually, the primary focus of early-stage companies is to get their product through the regulatory submission process. In that case, some of the first QMS processes that a company should implement are:
-
Document control and records management
-
Risk management
-
Supplier management
Start there and worry about the other processes (CAPA, complaint handling, etc.) when you are closer to manufacturing and product launch.
Implementing a QMS and keeping it updated can feel overwhelming, but the degree of difficulty often comes down to the type of QMS solution you choose. Going with a paper-based QMS will create an enormous amount of manual work and ensure that there are missing records and signatures down the line. On the other hand, an extensive QMS software solution that requires customization to fit your needs will be time-consuming and difficult to set up and keep validated.
That’s why at Greenlight Guru, we built our QMS software to be the perfect solution for MedTech companies. Greenlight Guru Quality comes with compliant workflows and built-in traceability throughout the entire system, but it doesn’t take months to build out a customized workspace. Instead, you can quickly begin adding documents and working within the system to drive adoption and ensure your team’s success.
Before IVD manufacturer Hart Biologicals began using Greenlight Guru Quality, they were exhausted by their inefficient, paper-based system. The labor required to chase down documents, collect signatures, and deal with nonconformances was becoming overwhelming.
After switching from paper to Greenlight Guru’s QMS software, Hart Biologicals was able to cut audit prep time in half and eliminate all outstanding nonconformances, some of which had been open for years. Best of all, Greenlight Guru helped accelerate their development process by 75%, turning an area full of delays into a series of efficient workflows.
Start with feasibility testing
Depending on where the device is in its development, the early testing you do will be to prove whether the idea for your device is even feasible. Feasibility testing, if it is conducted without the use of human subjects or human tissue samples, generally does not require FDA oversight.
Below is an example of the testing progression that you might go through in bringing your IVD to market.
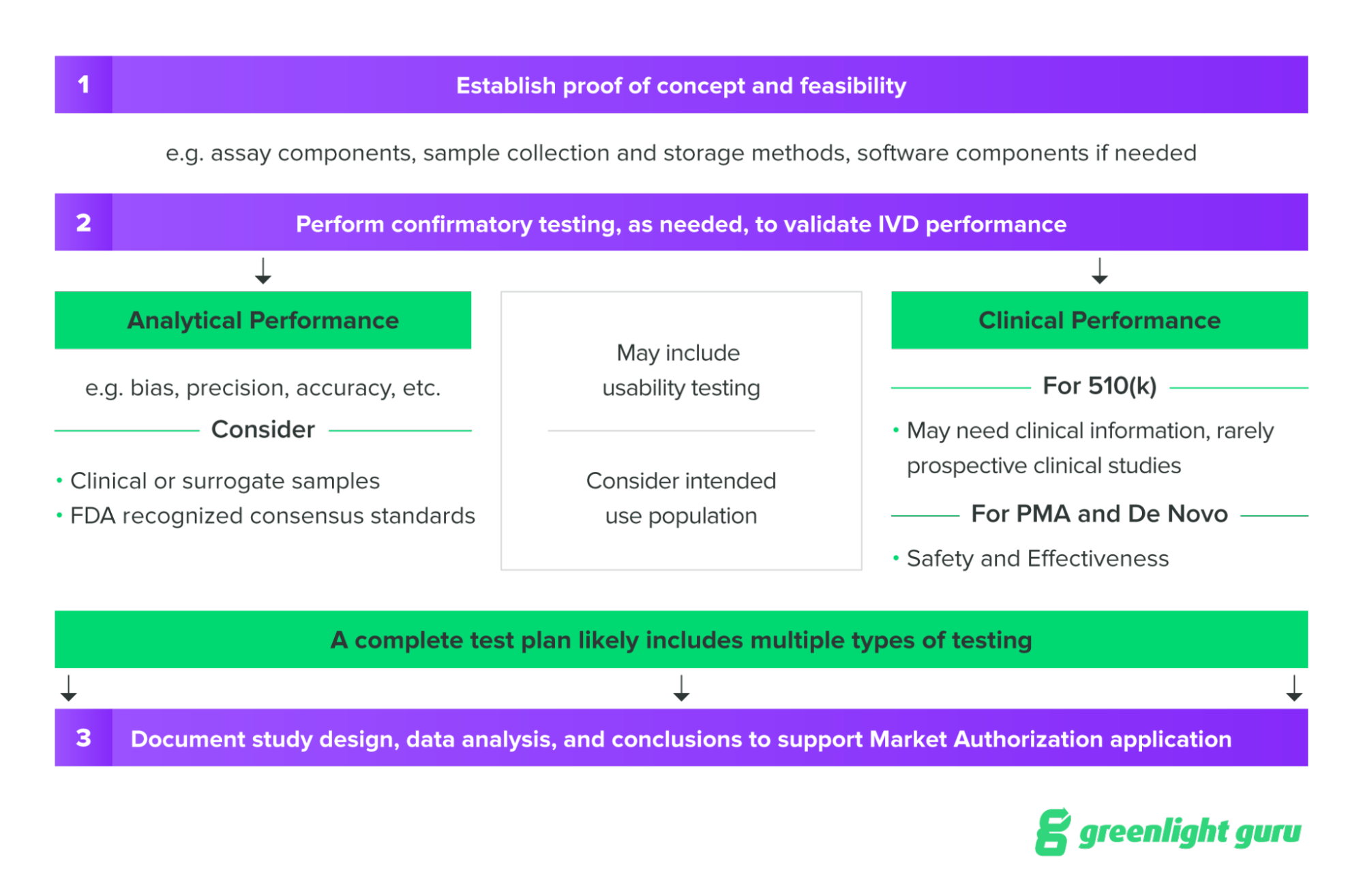
Source: NIH SEED Regulatory Knowledge Guide for In Vitro Diagnostics
Before feasibility testing, one of the first steps you’ll need to take is establishing the basic performance characteristics of your device, in order to plan confirmatory testing later on. Some of the performance characteristics to establish prior to confirmatory testing may include:
- Accuracy is the closeness of a measurement to the true value
- Precision is closeness of repeated measurements to one another
- Reportable range is the range of analyte concentration for which the test system can accurately report results
- Reference intervals are the range of IVD output values that correspond to normal or healthy populations
Two of the most important characteristics are sensitivity and specificity, which determine the accuracy of the test. The sensitivity of a test refers to its ability to yield a positive test result when a patient has the disease or condition being tested for. Specificity, on the other hand, refers to the ability of a test to provide a negative result for a patient who does not have the disease or condition.
Sensitivity and specificity tend to be inversely related. So a highly sensitive test that is more likely to produce a positive result will typically have less specificity. Both types of tests—highly sensitive and highly specific—have their uses. A highly sensitive test may be used to ensure that a certain condition is not missed in screening. And a highly specific may be used to more definitely rule out a disease or condition.
Given that the risk of false negatives or false positives is essentially how risk class is determined for IVDs, it’s important to have a deep understanding of the intended use of your device and both its sensitivity and specificity.
Perform analytical and clinical validation
Two major points on the journey to market are the analytical validation and the clinical validation of your device. This validation is the confirmatory testing I mentioned in the previous section.
Analytical validation refers to demonstrating the technical reliability of your IVD. In other words, how well does it measure the analyte you’re targeting?
We’ve already discussed one method of analytical validation, which is comparing your device’s performance to a predicate device. This is commonly used for devices taking the 510(k) pathway to market, and it requires you to demonstrate that your device is at least the same in all capacities to support a claim of substantial evidence.
When designing analytical validation studies, you’ll want to consider several important factors that could impact your device’s accuracy and reliability. These can include:
- Factors of uncertainty. Your validation plan should include all factors that could affect diagnostic performance and account for as many as possible.
- Limitations of measurement. Identify which metrics—such as reference interval, limit of detectability, sensitivity/specificity, or linearity—are applicable to your IVD and determine optimal operating thresholds. You’ll also want to identify the upper and lower limits of those thresholds.
- Accuracy and precision. Your analytical test plan will likely need to include a study to determine the accuracy and precision of the device’s measurement. Accuracy measures the amount of bias in the IVD, while precision measures the repeatability and reproducibility of the IVD.
Clinical validation is intended to show that the measurements of your test are capable of reliably identifying, measuring, monitoring, or predicting the presence or absence of a clinically defined condition, disorder, or health status in a clearly defined target population.
As previously mentioned, an analytical performance test comparing your device to a predicate device is often sufficient for demonstrating clinical validity. However, if your IVD is using novel or unproven technology, you may need to use a prospective clinical study to demonstrate clinical validity, as well as safety and effectiveness. This is typically the case with devices that are using the PMA or De Novo pathways in the US.
If you do need to perform a clinical study it’s a good idea to think carefully about how you’re capturing and managing data for that study. Given the importance of clinical data in your regulatory submission, you need an electronic data capture (EDC) system that makes building studies and capturing high-quality and reliable data as easy as possible.
That’s why at Greenlight Guru, we built our EDC system specifically for MedTech companies that need a modern, compliant, and easy-to-use EDC system that fits their unique clinical needs. Greenlight Guru Clinical comes with built-in compliance with both FDA and EU regulations, as well as ISO 14155:2020.
Prepare for post-market surveillance
Whether you’re in the US or the EU, you are required to follow your product throughout its entire lifecycle, collecting data on the product while it’s on the market. The point of post-market surveillance (PMS) is to quickly discover safety issues with the device and more accurately understand how it’s being used on the market.
While people often think of complaint handling or nonconformance management when they think of post-market surveillance, your PMS program should also be used to potentially improve the product and inform new iterations of the device. It can help you answer questions like:
-
Is our device too difficult to manufacture?
-
Do we need to change materials?
-
Do we need to update our IFU or labeling?
-
Do we need to change a supplier who isn’t meeting our standards of quality?
-
Can we market our device differently, using post-market clinical evidence?
-
Can we use reimbursement pathways we didn’t before?
If you decide to implement only the essential parts of your QMS during product development, you’ll need to establish QMS processes related to post-market surveillance before your device goes on the market. These include:
-
Nonconformance management
-
Corrective action and preventive action (CAPA)
-
Internal auditing
If used correctly these processes and the data they generate should provide you with a strong foundation for postmarket surveillance. Remember, you need to start early and be proactive when it comes to post-market surveillance. This is especially important if your device is in the EU and you need to comply with IVDR’s PMS requirements.
One of the best pieces of advice here is to build a strong chain of communication for complaints and feedback. The existence of a complaint is no guarantee that something will be done about it. It’s easy for complaints coming from the field—by a sales rep, for instance—to get siloed or lost. And if that happens often enough, not only will you miss potentially serious issues with the product, you’ll incentivize your reps to stop sending in complaints.

Streamline your IVD journey with a MedTech-specific solution
As you can see, there’s a lot to consider on the journey to getting an IVD to market. And given that you’ll already be making crucial decisions regarding design and development, clinical data collection, regulatory pathways, and more, you need software solutions that simplify it for you.
At Greenlight Guru, we’ve built our quality management and electronic data capture solutions specifically for MedTech companies, because we know how unique this industry is. We know that using paper or pharma-specific solutions simply doesn’t work for MedTech. That’s why we created Greenlight Guru Quality and Greenlight Guru Clinical with your needs in mind—to make it easier to get life-changing devices to market faster.
When you choose Greenlight Guru, you get modern solutions with built-in compliance to FDA and EU regulations, as well as standards like ISO 13485:2016, ISO 14971:2019, and ISO 14155:2020. On top of that, you’ll get hands-on guidance from experienced MedTech professionals who have been in your shoes and can advise you on regulations, risk reduction, business roadblocks, and much more.
See what purpose-built software can do for your MedTech company by getting your free demo of Greenlight Guru →
FREE eBOOK:
Ultimate Guide to In Vitro Diagnostic (IVD) Devices
Ultimate Guide to In Vitro Diagnostic (IVD) Devices

%20Ultimate%20Guide%20to%20In%20Vitro%20Diagnostic%20(IVD)%20Devices.png?width=1700&name=(cover)%20Ultimate%20Guide%20to%20In%20Vitro%20Diagnostic%20(IVD)%20Devices.png)
%20Ultimate%20Guide%20to%20In%20Vitro%20Diagnostic%20(IVD)%20Devices.png?width=250&height=324&name=(cover)%20Ultimate%20Guide%20to%20In%20Vitro%20Diagnostic%20(IVD)%20Devices.png)



