
FDA has officially announced their Final Rule regarding the phaseout of enforcement discretion for laboratory developed tests (LDTs).
These tests will now fall under the same enforcement approach as FDA takes with other in vitro diagnostics (IVDs), which FDA believes will better protect the health and safety of patients who rely on the results of LDTs.
(The Final Rule will be published in the Federal Register on May 6, 2024, but a PDF version is available leading up to that date.)
This is a huge change for the labs that make LDTs. And while it has been in the works for years, it’s essential that everyone affected understands the fundamentals of the new enforcement framework and how it will impact their work. So in this article, I’m going to lay out the basics of the rule: what’s changing and how the change will be implemented.
Stay tuned, though, as we’ll be following this up with advice on the way forward for LDT manufacturers who will soon need to comply with IVD regulations.
LDTs vs IVDs: what’s changing and why?
Until now, the difference between LDTs and IVDs, from a regulatory standpoint, was who manufactured them. IVDs are produced by device manufacturers who must comply with the FDA’s premarket and postmarket controls.
LDTs, on the other hand, are manufactured by laboratories, and (until now) fell under the jurisdiction of the Center for Medicare and Medicaid Services (CMS). Previously, they were subject to a different set of regulations than IVDs, which are known as the Clinical Laboratory Improvement Amendments (CLIA).
However, FDA believes this regulatory scheme—with IVDs and LDTs regulated differently—is not in the best interest of public health. In the announcement, they state that most modern LDTs pose a greater risk than they did decades ago, when FDA originally decided on its policy of enforcement discretion.
The agency also notes that they are “aware of numerous examples of potentially inaccurate, unsafe, ineffective or poor quality IVDs offered as LDTs that caused or may have caused patient harm, including tests used to select cancer treatment…”
(We’ve covered more of the background for the rule change in previous articles and podcast episodes, if you’re interested.)
And due to those public health risks that FDA lays out in the Final Rule (Chapter III, Section B), they have decided to end enforcement discretion and bring LDTs under the same regulatory umbrella as other IVDs.
What does the amendment in the LDT Final Rule actually say?
Despite the considerable length of the Final Rule, the amendment that changes the way LDTs are regulated is quite short.
FDA is amending its regulations to update the definition of “in vitro diagnostic products.” The regulations, such as 21 CFR Part 809, subpart A, will now state that IVDs are considered devices under the FD&C Act including when the manufacturer of the IVD is a laboratory. That, however, is enough to bring LDTs under FDA jurisdiction, and much of the Final Rule is given over to explaining exactly how that will work.
For those wondering how this will affect the availability of tests during extreme situations, FDA also released two draft guidances along with the Final Rule. The first draft guidance covers the agency’s thinking regarding enforcement discretion policy for labs offering unauthorized IVDs in response to an emergent situation, like the outbreak of an infectious disease. The second draft guidance offers insight into the factors FDA will consider when developing public policy for enforcement discretion for IVDs during a public health emergency.
When will LDTs be subject to FDA’s IVD regulations?
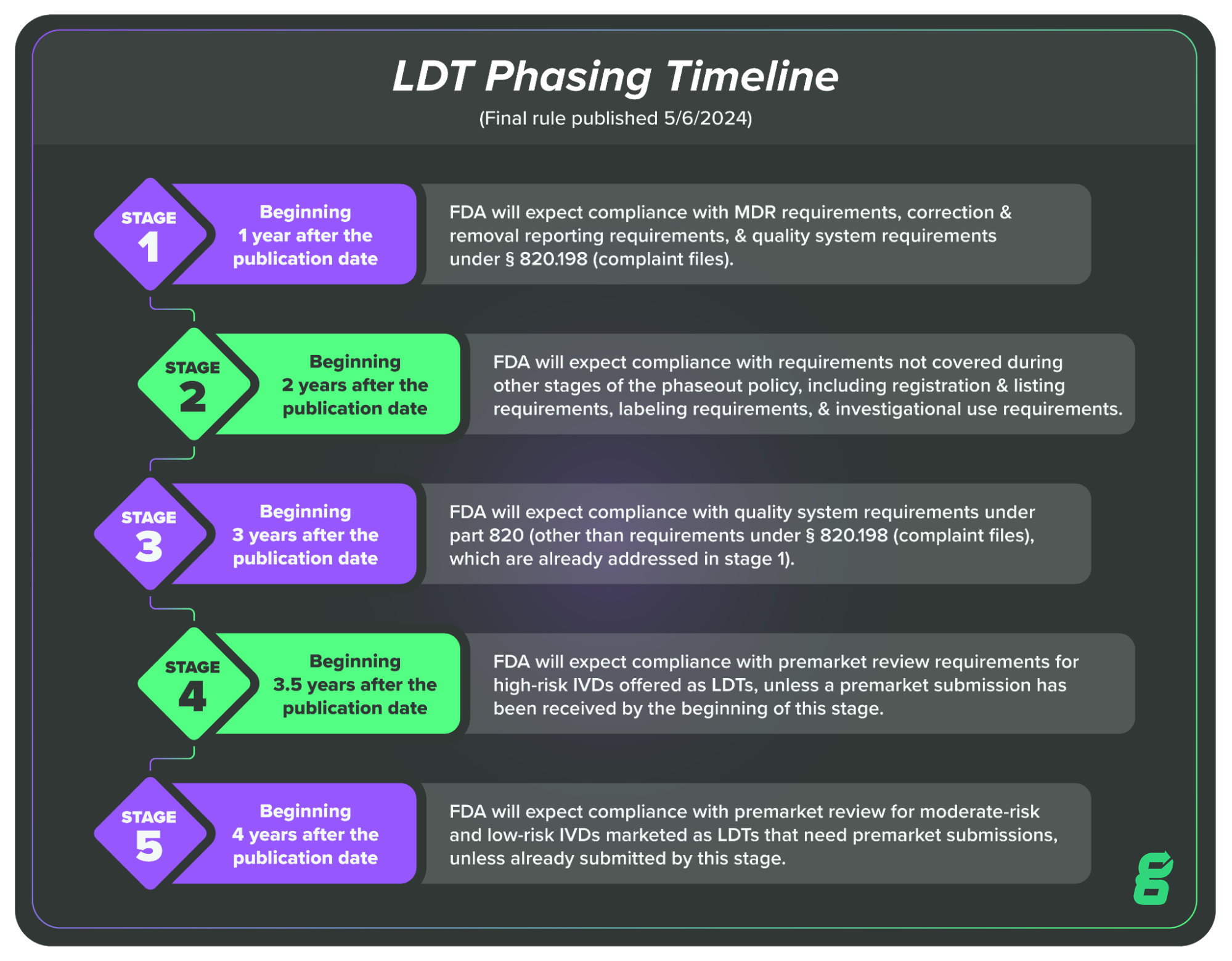
Chapter V of the Final Rule, Phaseout Policy describes how FDA plans to phase out their “enforcement discretion” and when those phases will occur. This essentially means FDA is phasing in the enforcement of IVD regulations for LDTs, giving LDT makers more time to hit more difficult regulatory compliance targets.
This phasing in period will take four years, at the end of which all LDTs will be expected to meet the relevant regulatory requirements for IVDs. The four-year period will be broken up into five key stages:
-
Stage 1: Beginning 1 year after the publication date of the final rule, FDA will expect compliance with MDR requirements, correction and removal reporting requirements, and quality system requirements under § 820.198 (complaint files).
-
Stage 2: Beginning 2 years after the publication date of the final rule, FDA will expect compliance with requirements not covered during other stages of the phaseout policy, including registration and listing requirements, labeling requirements, and investigational use requirements.
-
Stage 3: Beginning 3 years after the publication date of the final rule, FDA will expect compliance with quality system requirements under part 820 (other than requirements under § 820.198 (complaint files), which are already addressed in stage 1).
-
Stage 4: Beginning 3½ years after the publication date of the final rule, FDA will expect compliance with premarket review requirements for high-risk IVDs offered as LDTs, unless a premarket submission has been received by the beginning of this stage, in which case FDA intends to continue to exercise enforcement discretion while its review is pending.
-
Stage 5: Beginning 4 years after the publication date of this final rule, FDA will expect compliance with premarket review requirements for moderate-risk and low-risk IVDs offered as LDTs (that require premarket submissions), unless a premarket submission has been received by the beginning of this stage in which case FDA intends to continue to exercise enforcement discretion while review is pending.
While the phasing in of enforcement will take four years, if your lab makes LDTs, now is the time to begin preparing to meet these requirements. In our follow-up article, we cover Stage One of the LDT Enforcement Discretion Phaseout, including medical device reporting requirements, which is the first big step in enforcement.
Greenlight Guru is here to help you meet all your IVD regulatory requirements
At Greenlight Guru, we’ve been helping medical device manufacturers stay compliant with regulations, pass audits, and deliver safe and effective devices to patients faster for more than a decade. As FDA phases out enforcement discretion of LDTs, your lab will need to begin meeting regulatory requirements for devices, such as premarket review and quality system requirements.
No one makes meeting these requirements simpler and easier than Greenlight Guru. Whether you need a compliant, modern QMS solution or a clinical data collection system for upcoming clinical trials related to a premarket submission, our MedTech Suite has you covered.
We’re about to enter a period of enormous change for laboratories producing LDTs, but that doesn’t mean you have to face these changes alone. Contact us to learn more about how we can help you meet your IVD regulatory requirements by getting your free, personalized demo today →
Free eBook: Medical Device Classification Guide






