
On May 6, 2024, FDA published their Final Rule marking the end of enforcement discretion for lab developed tests (LDTs).
Federal regulations will now state that in vitro diagnostics (IVDs) are considered devices subject to FDA regulations including when the manufacturer is a laboratory.
This is a huge shift in the regulatory landscape for LDTs, which were previously subject to a different set of regulations, known as the Clinical Laboratory Improvement Amendments (CLIA). Given the magnitude of the change, FDA has laid out a four-year, five-stage transition plan to phase out enforcement discretion (or phase in enforcement).
Here’s what the LDT transition looks like in brief:
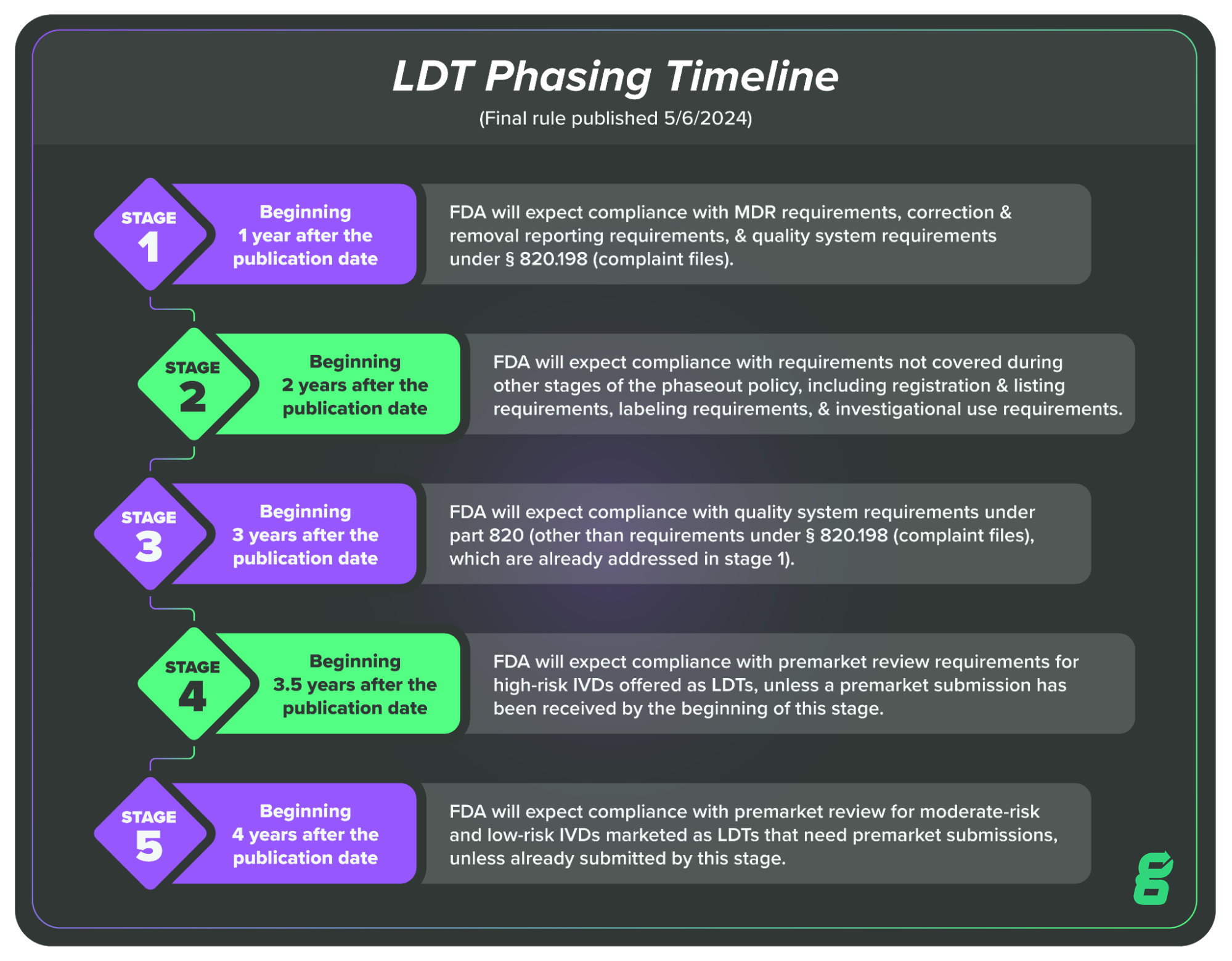
In this article, we’ll cover Stage One, which LDT manufacturers will be required to comply with one year after the publication of the final rule, meaning May 6, 2025.
BONUS RESOURCE: Click here to download a free, customizable Complaint Form Template.
What is required in Stage One of the LDT timeline?
Here is what FDA states will be required in Stage One of the enforcement discretion phaseout:
Beginning 1 year after the publication date of this final rule, FDA will expect compliance with MDR requirements, correction and removal reporting requirements, and QS requirements under § 820.198 (complaint files).
FDA’s reasoning here is that they believe some IVDs offered as LDTs may be posing a risk to patients (via false negatives, for example) and they want to begin getting information on those LDTs as quickly as possible.
This is also why FDA is prioritizing requirements on correction and removal information. This type of reporting goes hand-in-hand with MDR requirements, as manufacturers initiate correction or removal of IVDs when there is a risk to patient health or in order to remedy a violation of the FD&C Act.
Finally, FDA has also included the requirement that LDT manufacturers begin complying with the Quality System requirements for complaint files (21 CFR Part 820.198). This also works together with the MDR requirement because manufacturers need to be able to document and investigate complaints regarding their products in order to determine if they are required to report the issue under MDR requirements.
In essence, we can think of Stage One of the enforcement discretion phaseout as an attempt to get more information on LDTs that may be posing risks to patients. Each of the requirements is related to the others, and all are moving toward the same goal of surfacing information on potentially harmful tests.
How can labs comply with MDR and the other requirements in Stage One?
LDT manufacturers only have one year to become compliant with the requirements of Stage One, so let’s look at each of the three requirements and the regulations you’ll need to understand as you work toward compliance.
What is medical device reporting and who needs to do it?
The requirements for medical device reporting are detailed in 21 CFR Part 803. In the regulation, the FDA identifies three groups of mandatory reporters:
-
Medical device manufacturers
-
Medical device importers
-
Device user facilities
Each of these groups has separate and distinct requirements for reporting medical device adverse events. Since LDT manufacturers now fall into the category of device manufacturers, they will be subject to that set of medical device reporting requirements.
Manufacturers must submit a report within 30 days of becoming aware of information from any source which suggests a marketed device may have caused a death or serious injury. The same is expected if the device experiences a major malfunction that could cause death or serious injury if it were to recur.
In general, manufacturers should use the mandatory reporting form, FDA Form 3500A. However, manufacturers may be required to submit a 5-day report when a reported event requires immediate remedial action, such as a product recall.
What are the Quality System requirements for complaints?
You can find the requirements for complaint handling in 21 CFR Part 820.198, which is part of the current Quality System Regulation (QSR). This portion of the QSR provides you with the requirements for receiving, reviewing, evaluating, and investigating complaints about your device, as well as record keeping.
In the QSR, FDA defines a complaint as, “any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution.”
As I noted earlier, in a certain sense, the requirements that FDA has used for Stage One give you a roadmap to compliance. By setting up quality system procedures for complaint handling, you’re putting yourself in a position to collect and investigate complaints that may be indicative of an MDR reportable event.
I’d highly recommend you check out our recent episode of the Global Medical Device Podcast with Lisa Van Ryn, in which she walks us through the relationship between MDR and complaints.
Finally, it’s important to point out that FDA published their Final Rule amending the QSR to better align with ISO 13485:2016. The effective date for enforcement of the new Quality Management System Regulation (QMSR) is February 2, 2026.
So, if you’re working on becoming compliant with Part 820.198, it’s a good idea to familiarize yourself with the ISO 13485:2016 requirements for complaint handling (Section 8.2.2) and control of records (Section 4.2.5).
If becoming compliant sounds daunting, know that Greenlight Guru’s QMS solution comes pre-validated per both FDA and ISO requirements. With dedicated complaint management software that’s connected to the rest of your QMS, you’ll have a single, up-to-date workflow to help you track and act on complaints in a timely and compliant manner.
What are the requirements for corrections and removals reporting?
You can find the requirements for corrections and removals in 21 CFR Part 806. Generally speaking, the regulation requires manufacturers to “report to FDA any correction or removal of a medical device if the correction or removal was initiated to reduce a risk to health posed by the device or to remedy a violation of the act caused by the device which may present a risk to health.”
However, if the information about a correction or removal has already been reported to FDA through MDR, the manufacturer is not required to make a report. You’ll need to make the report to FDA within 10 days of the correction and removal.
If the reason for the correction or removal does not pose a “risk to health,” you’ll still need to document it and keep it for your records, but you are not required to file a report.
BONUS RESOURCE: Click here to download a free, customizable Complaint Form Template.
Comply with FDA regulations during and after the enforcement discretion phaseout with Greenlight Guru
If you’re unfamiliar with the QSR or any of the other documentation and reporting requirements that you’ll need to comply with in Stage One (and beyond), the enforcement discretion phaseout may feel overwhelming.
Fortunately, you don’t have to do this alone. At Greenlight Guru, we’ve been helping MedTech companies stay compliant with regulations, pass audits, and deliver safe and effective devices to patients faster for more than a decade. As the phaseout continues, you’ll need to go on building a compliant quality system and ensuring you have a single source of truth for all your records.
No one makes meeting these requirements simpler and easier than Greenlight Guru. Whether you need a compliant, modern QMS solution or a clinical data collection system for upcoming clinical trials related to a premarket submission, our MedTech Suite has you covered.
Discover how Greenlight Guru can help you meet your IVD regulatory requirements by getting your free, personalized demo today →
Complaint Form Template










