
The purpose of any medical device is to benefit others and improve the quality of life. However, in order to keep patient and user risk at a minimum, authorities have tightened up regulations over recent years with the hope that manufacturers will build quality in, while taking risk out of a device.
However, trying to navigate the product lifecycle journey alone can be tricky, even intimidating. That’s why we created the following flowchart “Concept to Market'' diagram to guide early stage medical device manufacturers when developing a new medical device.
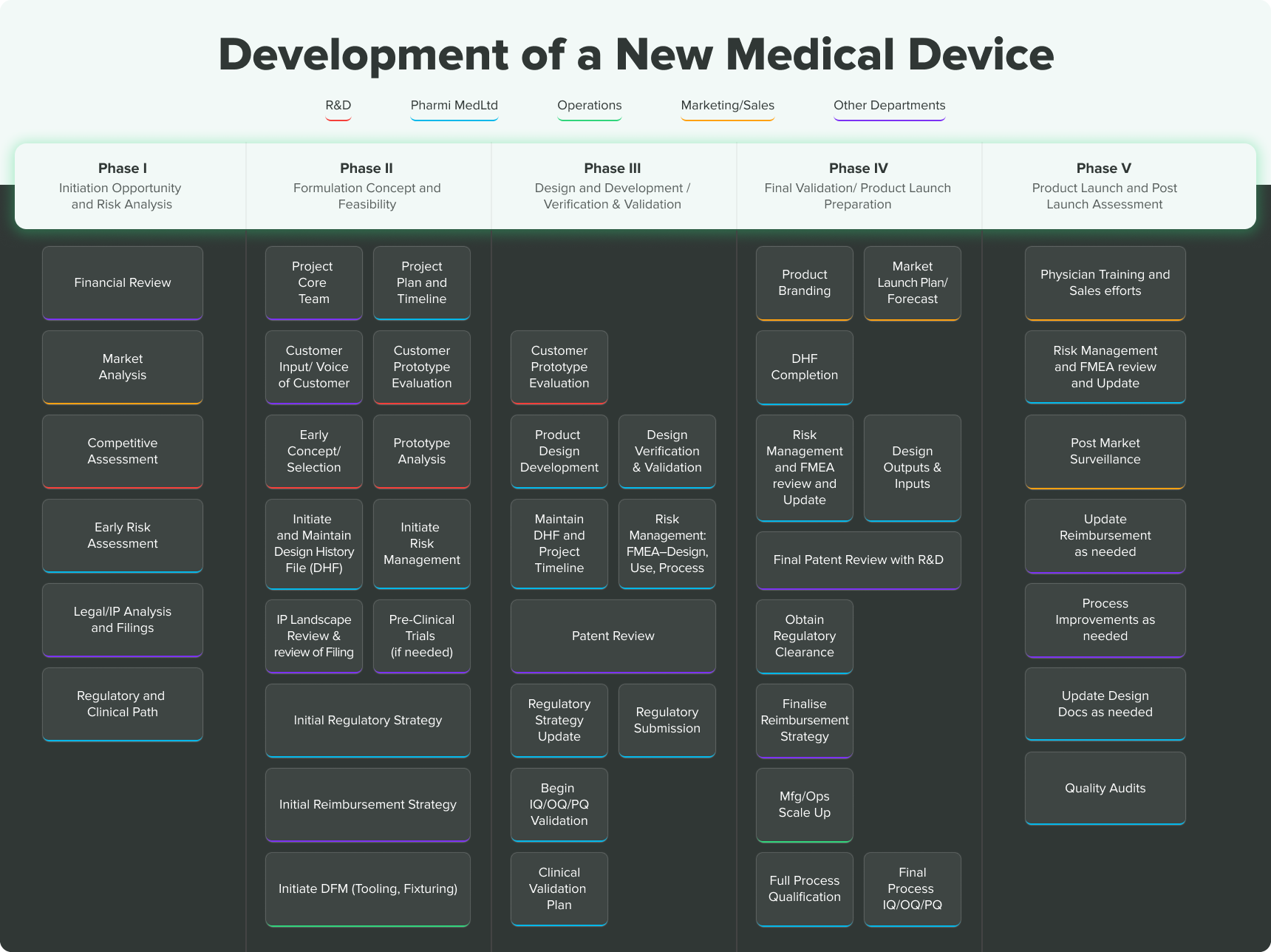
Keep in mind, the steps and stages are not set in stone. This is meant purely as a general overview of what obstacles and opportunities may lie ahead. That said, there are 5 phases of medical device development you need to know about, as well as some general best practices to follow.
5 Phases of the Medical Device Development Process
Medical Device Development Phase I – Initiation, Opportunity and Risk Analysis
The first phase of the medical device development process is all about planning, researching, and a whole lot of documentation.
Think about all the work that will be involved in the product development process. Think of the highly regulated medical device industry. Consider your capabilities for medical device manufacturing. And admit that you will not be able to tread this path alone.
Think about your funding strategies, which markets you want to enter – where? Why? and How? This is fundamental as each market has its own regulations and challenges. If you are developing a new product that no one has produced before, make sure you protect your IP (intellectual property) and consider patenting your idea.
Your market research is fundamental as it will lead to the following – are there equivalent devices on the market you can claim as a predicate? Or do you need to conduct clinical trials for your device? The difference between the two can make your venture feasible or infeasible due to the high costs involved. Don’t forget to document this.
Another foundational step in this first medical device development phase is implementing a quality management system (QMS). Your QMS is the foundation to your company, which once solid will provide a good base on which you can build.
ISO 13485:2016 is the standard that is adopted by most medical device companies and is internationally recognized, so get this in place. Your QMS will incorporate your procedures, forms, templates, and SOPs for how you will lock down and control all activities related to your medical device.
Medical Device Development Phase II - Formulation, Concept and Feasibility
Phase II is all about developing the concept and proving that it works. This is the best time to start thinking about risks and customer requirements. The customer’s voice is crucial, so be sure to solicit feedback via surveys and interviews. Use these insights, along with the competitor analysis and market research, and adopt this into the design of your product.
Once you have a proof of concept and are convinced that your device has a market position, is viable and financially feasible, only then should you begin to seek funding for prototyping and trial runs on your device.
Additionally, make sure you know the regulatory requirements for your device, as well as the regulatory and safety requirements of the country/region you plan to sell in (which should have been determined in your market research). The requirements in the US differ from those in Europe, and although there is very strong overlap, the product submission processes are quite different.
Medical Device Development Phase III - Design & Development, Verification & Validation
Now your device is beginning to take shape—you have a prototype, and have done some trials, but you still haven’t really put the design through its paces. That’s why the third medical device development phase focuses on validation and verification of your device as a way to prove it will indeed withstand all the pressures of the real world.
Make sure you know what your acceptance criteria are for each test. A good way of doing this is by setting up a design traceability matrix, which will ensure you have not lost track of your customer requirements. You will need to translate this into engineering requirements, which answer questions such as:
-
How will you fulfill the customer requirement?
-
What mode of testing will you use to verify and validate?
-
What processes do you need?
-
What test equipment will you need?
-
Have you started to think about a manufacturing and quality plan, or will you outsource this?
This is also an opportunity to lean on your risk management practices. Make sure you document all the potential failures of what can go wrong due to bad design, poor manufacturing process, or user failures by foreseeable misuse. Be realistic and think this through. I cannot emphasize doing this thoroughly enough, as your device will not make it to market if the device is seen to be a risk to patient safety.
Don’t forget to make any necessary updates to your regulatory strategy. If you had determined earlier that you needed clinical trials for your product, you should begin your clinical investigation and use the product for the trials from your design freeze through actual use in a clinical environment. Also look at what external approvals you will need, such as an ethics committee.
Medical Device Development Phase IV - Final Validation and Product Launch Preparation
As we enter the fourth phase of medical device development, It’s time to start thinking about marketing and branding. One word of caution—be careful what you put in your marketing literature. If you’re making a claim, it must be backed up by evidence. It makes me cringe when I see marketing experts make claims on products that have no justification.
Furthermore, regulatory agencies like the FDA consider marketing materials to be a form of labeling, and have strict requirements for it in its Quality System Regulation (QSR), 21 CFR Part 820. My message is no evidence = no claim.
All too often I see companies get to this stage of medical device development only to realize they didn’t finish all the required testing. Why? Because they didn’t know the regulatory requirements for the product, took shortcuts or didn’t see the real reason to do the test.
You should be gathering all data for validation and verification tests, as well as documenting your evidence of testing, such as biocompatibility, electrical safety, stability, and shipping.
You should also be completing your technical documentation at this stage. Create your tech file full of all the evidence required to put in front of a competent authority that will review your file for completeness against expectations.
Medical Device Development Phase V - Product Launch and Post-Market Surveillance
Before you begin celebrating an upcoming product launch, it’s important you have a final review with your team about some can’t-miss details.
-
Is your technical documentation complete and current?
-
Do you need to re-test?
-
Are your post-market plans in place?
-
Do you have a system in place to capture complaints and feedback?
-
Have you finalized packaging and labeling?
-
If in Europe, do you need to report any significant changes to the competent authorities?
OK, guess what? I think you could be ready now! If your product submission and technical file review is done, you are ready to launch your product. Once you have your license or certificate you can place your device on the market.
Your device is now in the hands of users so be ready for customer feedback, improvements and continuously reviewing your internal systems. Make sure you have a robust internal quality auditing system to review your records and ensure you are:
-
Not cutting corners in medical device production
-
Following your QMS procedures
-
Adhering to good manufacturing practices (GMP)
-
Following your feedback and complaints system
-
Updating all necessary technical documentation as required
-
Adequately resourced
-
Continuously improving medical device design
Get safer products to market, faster with Greenlight Guru
Bringing a medical device to market can be both financially rewarding and, more importantly, improve patient lives. By no means should the process be rushed as the authorities will be watchful for vigilance to ensure you’re following regulations and complying with requirements as necessary.
At Greenlight Guru, we want you to do more value-added engineering work and less compliance paperwork. Our end-to-end medical device development solution accelerates your product development efforts, allowing you to drive collaboration, unlock traceability, and gain visibility into critical information throughout your product’s lifecycle.
And at any stage of your commercialization journey, we enable you for success with our medical device QMS software and support you in your journey of bringing life-changing solutions to patients. Ready to learn more? Contact us today for your free, personalized demo of Greenlight Guru →
FREE RESOURCE:
Ultimate Guide To Design Controls for Medical Device Companies
Ultimate Guide To Design Controls for Medical Device Companies

%20Design%20Controls.png?width=2550&name=(cover)%20Design%20Controls.png)
%20Design%20Controls.png?width=250&height=324&name=(cover)%20Design%20Controls.png)



