





Getting a medical device to market is a huge accomplishment for any MedTech company. Design and development, regulatory approval, supplier management and manufacturing, marketing and sales (to name a few) all have to be working in concert to put a device in the hands of an end user.
But once a medical device is on the market, that doesn’t mean the work is over. Not by a long shot. There are your post-market regulatory requirements, like the various post-market surveillance activities you’ll be required to perform. Then there’s manufacturing and supplier management to think about.
Keeping up with the market as it evolves, expanding your product portfolio, or adding new intended uses to your device—they’re all part of successfully managing a device once it’s on the market.
That’s why we’ve created this guide for MedTech companies that are managing devices already on the market. It’s a wide-angle look at the various responsibilities and considerations that revolve around managing a device on the market.
When you need a holistic overview of the many facets of managing a medical device after market entry, this guide will be a resource you can return to again and again.
So let’s dive in.
Table of Contents
|
Manufacturing & Supplier Management: Streamlining Production Streamlining And Scaling Manufacturing |
|
Ongoing Activity: Keeping Your Device on the Market Planning For Post-Market: Setting Yourself Up For Success |
|
Growth & Expansion: Increasing Your Device’s Reach New Indications For Use |
| End of the Line: Retiring a Device |
| From Design & Development to Obsolescence, Greenlight Guru Has Your Back |
 Manufacturing & Supplier Management: Streamlining Production
Manufacturing & Supplier Management: Streamlining Production
Your manufacturing processes and supplier relationships are two critical areas of managing your medical device while it’s on the market—and they’re also areas where you’ll be consistently trying to identify and stay ahead of any issues. According to one report, manufacturing defects were the leading cause for product recalls going into 2023.
1. Streamlining and scaling manufacturing
Planning is critical when it comes to getting the manufacturing of your device right, and using good project management principles will be key to your success.
When you first begin manufacturing your device, you’ll quickly learn that it’s one thing to set up your processes, procedures, and work instructions at your manufacturing facility. It’s another thing entirely to see how those processes play out in reality.
You may discover that a task takes longer than you thought, or that there’s plenty of room to make a process more efficient. In other words, you’ll almost certainly need to continually refine those processes and procedures until you get it right.
At this stage, you’ll be trying to get to the bottom of questions like:
When you begin thinking about scaling manufacturing, you’ll go through a similar stage of planning and tweaking processes. You’ll need to work closely with your Product Development team and your Manufacturing engineers, as your processes may change as you begin to increase the number of devices you’re producing.
For example, let’s say you’re using injection molding for one of your parts. When you needed to make these parts six at a time, the “gate” through which the material was injected was in a specific spot on the mold that was most convenient at that time. But if you need 12 of those parts produced at a time, that could change how the molds should be positioned, where the gates should be placed, among other things.
Product Development and Manufacturing need to work closely on these changes. It’s even possible this will require a design change to the device. And at that point, you need to look at how your design controls may be affected by that change. How will that impact risk management?
All of this needs to be documented on a change order, and depending on whether there are changes to the device, you may need to perform verification and validation again, and document it in a Letter to File or even a re-submission.
In general, you’d be wise not to underestimate the amount of time you may need to spend planning, doing trial runs, and tweaking your manufacturing processes. You’ll have to consider resources, time constraints, the size of the project, and the endpoints. And of course, you’ll need to make sure you have the suppliers in place to get you the materials, parts, and components you need for your devices.
2. Supplier management
The most important thing to understand about your supplier management is that it needs to be a living process. Once you’ve qualified the necessary suppliers, placed them on your Approved Supplier List (ASL), and begun ordering from them, it can be tempting to let supplier management slide to the back burner.
But the reality is that things can change very quickly in your supply chain.
How you manage those changes will often come down to your supplier agreements and your relationships with your suppliers. Your supplier agreements are one of the most important tools for managing your supplier relationships. Each agreement should stipulate how issues like supplier concession requests, for example, will be handled. They should also include language that ensures you’re being notified of any changes in the materials, parts, or components you’re being supplied with.
You also need to have contingencies in place for worst-case scenarios, like a supplier going out of business unexpectedly.
-
Do you have an alternate supplier for any critical parts? If not, how long will it take to find one and add them to your ASL?
-
Do you have enough of this part to cover your needs if that process should take several months? If not, can you order more than you need to build up a backlog?
-
Can your current suppliers keep up with your growth projections? Will your supply chain be ready if you decide to enter a new market?
These are the kinds of questions you’ll want to have the answers to in order to avoid disruptions and any frantic scrambling with your supplier management.
Good communication with your current suppliers will always help you to avoid large issues with your supply chain. But it’s important that you do the work up front to qualify your suppliers using a risk-based approach and then continue to monitor them via scorecards, audits, and regular communication.
If you’re looking for more information on effective and efficient supplier management, then check out our ultimate guide to supplier management.
![enterprise-guide-managing-medical-device-keeping-device-on-market]() Ongoing Activity: Keeping Your Device on the Market
Ongoing Activity: Keeping Your Device on the Market
 Ongoing Activity: Keeping Your Device on the Market
Ongoing Activity: Keeping Your Device on the MarketAs with any long-term proposition, keeping a device on the market for years or even decades requires ongoing attention and management.
Think of it a bit like keeping up with the maintenance of your house. If you stop looking into issues and hope they’ll go away, you may have a period of relative peace and quiet—followed by an extremely expensive period involving a lot of contractors. On the other hand, with regular care and maintenance, you can avoid many critical problems and get ahead of issues as soon as you spot them.
Here’s what I’d suggest in terms of “regular maintenance” for your medical device while it’s on the market.
1. Planning for post-market: setting yourself up for success
Starting on the right foot—planning ahead and ensuring your post-market surveillance program is ready to begin collecting and managing data on your device and is compliant with the regulations in your market(s)—will help you avoid a lot of common problems while your device is on the market.
The best piece of advice I can give you, which will be true for practically everything you do to manage your device, is to create a project plan and use good project management principles.
Whether you’re attempting to scale your manufacturing or expand into a new market, creating a project plan with milestones, a timeline, and a work breakdown structure for getting it all done will make your life much easier and help ensure you actually achieve your goal.
Post-market surveillance and your QMS
The foundational post-market surveillance processes in your QMS are:
-
Complaint handling
-
Nonconformance management
-
Corrective Action and Preventive Action (CAPA)
-
Internal auditing
Ideally, you’ll have all of these in place and ready to use before the device hits the market. But if you’re new to the post-market portion of the device lifecycle, you should take the time to educate yourself on all of these processes and how and when to use each.
For instance, what should really trigger a CAPA? How should you put together a nonconformance report? You want to know the answers to those questions ahead of time, rather than figuring them out on the fly when post-market data starts rolling in.
Post-market regulatory requirements in the US
Proper preparation also means being familiar with the post-market surveillance regulations and their requirements in any market your product is in.
In the US, your post-market surveillance is mostly encompassed by the quality processes I just mentioned—nonconformance, complaints, CAPA, and internal auditing. However, there are other potential postmarket surveillance activities that FDA may require from your company beyond your quality processes.
These can be found in 21 CFR Part 822 - Postmarket Surveillance, and apply to class II and class III devices that meet certain criteria.
Post-market regulatory requirements in the EU
The EU has a much more structured and explicit postmarket surveillance program than the US. The requirements for postmarket surveillance are outlined in Chapter VII of EU MDR.
You must have the same quality system processes in place that we’ve already discussed (as required by ISO 13485:2016). But whereas those quality processes encompass most of your postmarket surveillance activities in the US, they’re just the beginning of what will be required of you in the EU.
You can find a more in-depth analysis of the different aspects of postmarket surveillance in the EU in our Ultimate Guide to Postmarket Surveillance, but the following table will help clarify your responsibilities based on device class under EU MDR.
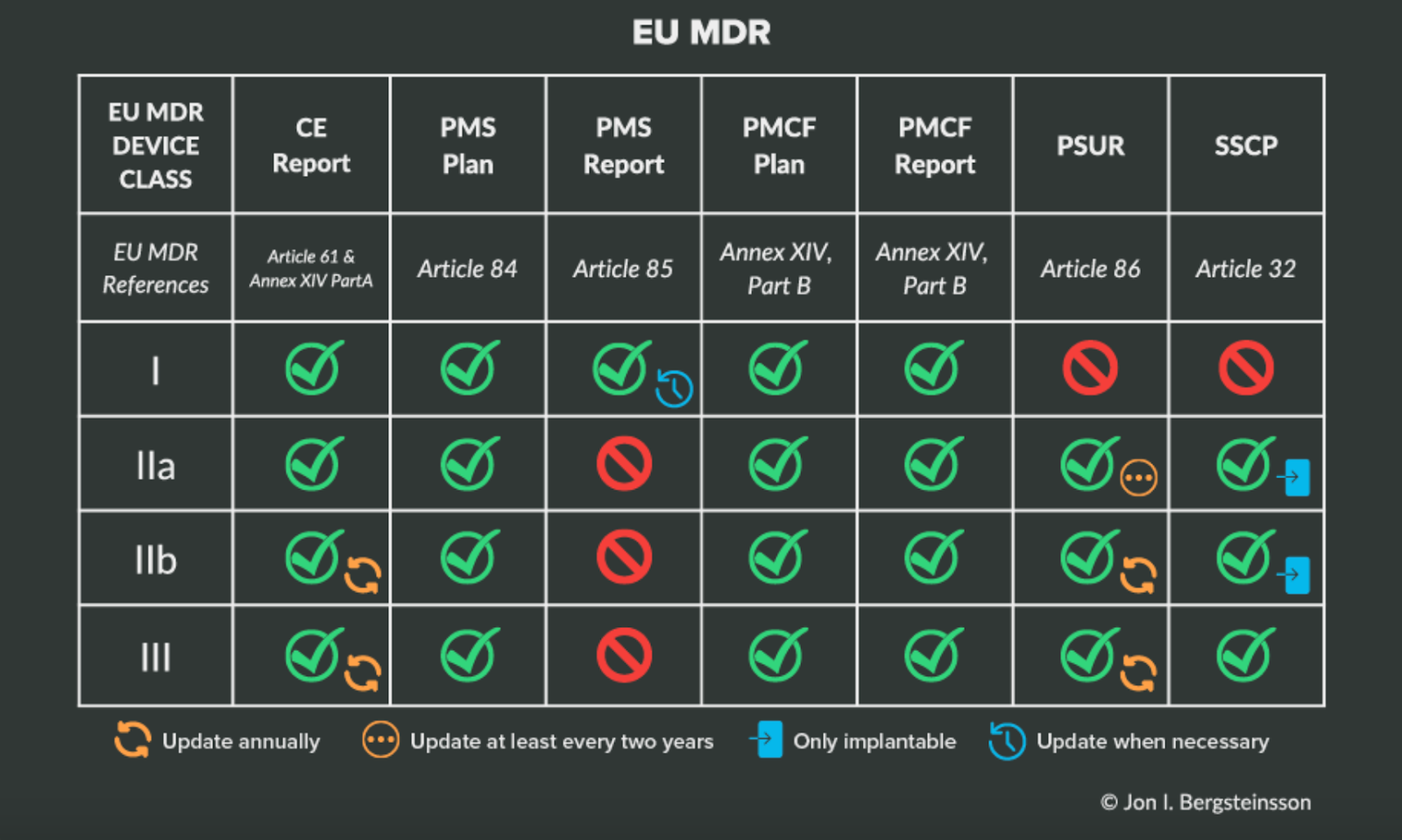
Again, the best thing you can do when it comes to post-market surveillance is to plan ahead. That work includes making sure you have a solution for collecting and managing clinical data from a variety of post-market sources, especially when it comes to compliance with the EU’s Post-Market Clinical Follow-Up or FDA Post-Approval Studies.
As the leading toolbox for MedTech clinical data collection, Greenlight Guru Clinical is the single, compliant platform for collection and management of all your clinical evidence, safety, and performance data. To learn more, get your free demo of Greenlight Guru Clinical today →
2. Managing post-market studies and data collection
In some cases, you may have studies that were started at the time your device was launched, and it’s critical that you keep up with them as they progress.
For instance, a lot of companies will need to collect real-time data into the aging of their device or its packaging. You are, of course, allowed to do accelerated aging testing to get a better understanding of how your device will age over the years.
But nothing beats reality, and you’ll need to follow along concurrently with a real-time study of your product and make sure you’re testing it at appropriate intervals to support the conclusions from your accelerated testing data.
Just because the accelerated tests say your packaging will last for five years, that doesn’t mean you can assert that timeframe and forget about it. It’s entirely possible that packaging that was found to last for five years in an accelerated aging test only lasts for three or four under real-world conditions.
3. Keeping up with the market for your device
Managing your own device is plenty of work, but I’d also encourage you to keep up with what’s going on in the market. What I mean by that is studying what’s happening in your device’s field and asking questions like:
-
What are our competitors doing? What’s the latest research emerging in fields that are related to our device?
-
When new clinical studies come out, what are they uncovering? Where do those results point?
-
What is state-of-the-art in this particular area right now?
It’s also important to keep up with any off-label use of your device. "Off-label" use refers to when a physician uses a legally marketed device outside of its labelling to treat a patient. While you can’t market your device for any off-label use, you should still have a good understanding of how this device is actually being used in the field. Knowing how clinicians are using your device can help you figure out which indications you may want to eventually market your device for once you have regulatory approval.
Knowing how your device is being used will also help you consider the next product(s) in your device portfolio. That data is incredibly valuable to you, and it’s essential if you want your device to have continued relevance in the market.
4. Modifying an existing device
There are a variety of reasons you may need to make a change to the design of your device during its time on the market. But whether it’s related to customer feedback, a change in materials or manufacturing processes, or something else entirely, a change to your device will always require documentation and may even require a new regulatory submission.
If you are considering a change, you’ll first want to consider what we call the three F’s: form, fit, and function. These refer to the identifying characteristics of the parts or components of your device:
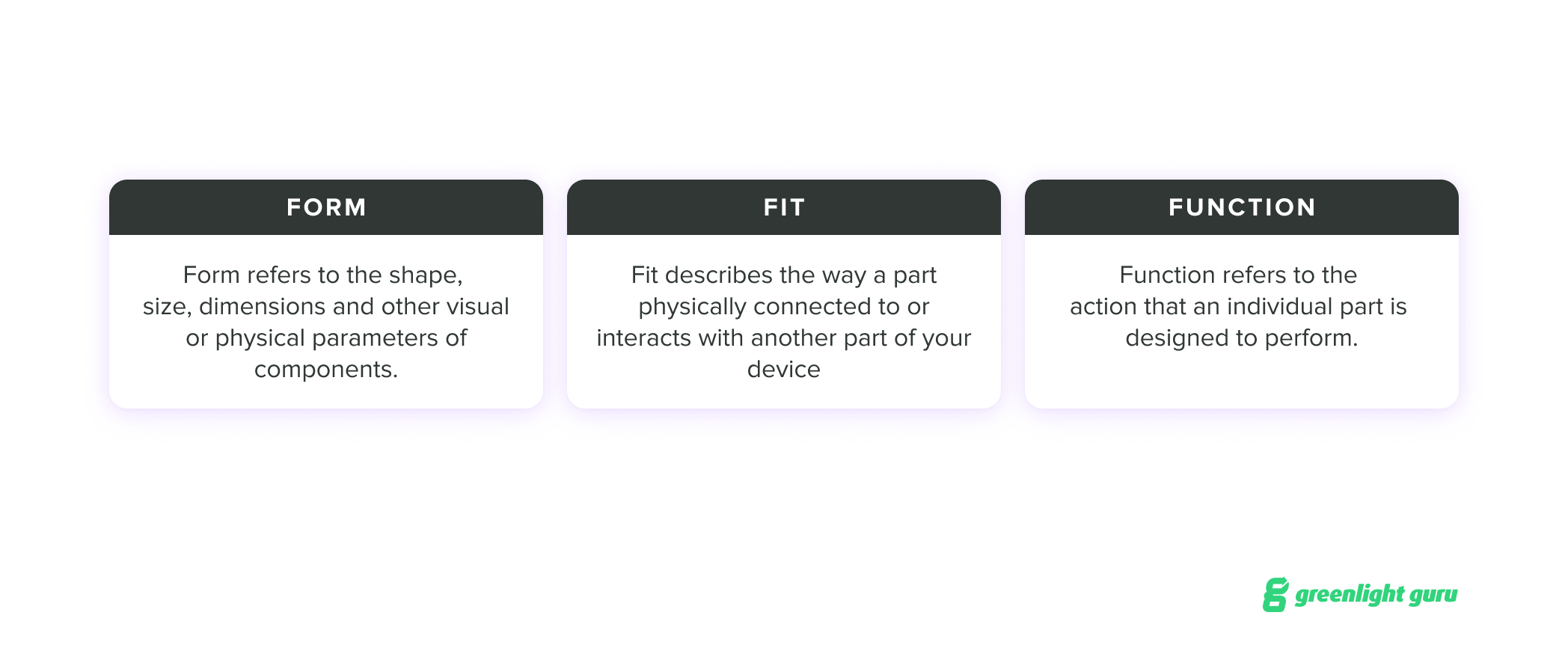
For a post-market change, you need to think in more granular terms to understand the impact your change might have on FFF when it comes to individual parts and their overall performance. Sometimes, a change may be trivial in these terms; other times, a single change may require additional changes to account for the impact on a device’s FFF.
For example, changing from one supplier to another requires due diligence to ensure that the new manufactured parts will be equivalent to the ones being replaced.
Remember, you should always conduct a change assessment when you’re modifying anything about a device, no matter how trivial it may seem. I’d recommend dedicating specific sections in your documentation to the change impact on form, fit, and function.
From a regulatory perspective, making a change may require you to complete additional regulatory documentation. In the US, devices may require a 510(k) submission, a post-market approval (PMA) supplement, or a letter to file. The FDA guidelines on change control can help you understand what’s required for any modifications you make to your device.
If you’re making changes to software as a medical device (SaMD) or software in a medical device (SiMD), FDA also has a guidance document on when to submit a new 510(k) for a software change to an existing device.
5. Understanding the evolving cybersecurity landscape
Many medical devices on the market today use software that can pose cybersecurity risks. If your device uses software or is software as a medical device (SaMD), it is incumbent upon you as the manufacturer to monitor, identify, and address cybersecurity risks as part of your post-market management of your medical device.
According to the FDA’s guidance document, manufacturers must implement comprehensive cybersecurity management programs and documentation consistent with 21 CFR Part 820, including (but not limited to):
-
Complaint handling
-
Quality audits
-
CAPA
-
Software validation and risk analysis
-
Servicing
The guidance document also includes an appendix that outlines the elements of an effective postmarket cybersecurity program.
While a comprehensive discussion of this topic is beyond the scope of this guide, you can find more information in our Ultimate Guide to Software as a Medical Device, as well as these episodes of the Global Medical Device Podcast:
-
Episode 264: Approaching Cybersecurity & Usability as a SaMD Company
-
Episode 270: Shifting Sands of SaMD Cybersecurity Regulations
6. Training and personnel management
There’s a theory out there about the “power of three” that posits that every time you increase the number of people in an organization by 3x, it typically entails large-scale change to your processes and management.
I bring this up because training management is a major part of the way your company operates and meets regulatory requirements. And the way you handle training when there are 100 people in the company may be very different from the way you do it when there are 300 or 900.
It’s important to reevaluate existing processes as you grow, both for the sake of ensuring that everyone is capable of doing their job and being able to prove it during an audit.
Once your company gets to a certain size (maybe one of those “power of three” moments), you may need to reassess how you’re proving that everyone has been properly trained. Does that mean quizzing? Or implementing a feedback system? Or conducting a 360 review where peers are evaluating each other?
Whatever you do, it’s essential that you’re regularly evaluating your training processes and ensuring that they’re effective—especially if your company is growing and changing.
![enterprise-guide-managing-medical-device-increasing-increasing-device-reach]() Growth & Expansion: Increasing Your Device’s Reach
Growth & Expansion: Increasing Your Device’s Reach
 Growth & Expansion: Increasing Your Device’s Reach
Growth & Expansion: Increasing Your Device’s ReachOnce your device is on the market, part of your strategy for growing and expanding its footprint will involve change—maybe to the device itself, maybe to the geographic area where you sell it, or maybe additional indications for use. So let’s take a look at some of the more common scenarios.
1. New indications for use
One method for growth when a device is on the market is adding new indications for use. A device's indications for use are simply the circumstances or conditions under which the device will be used. Indications for use will answer questions like:
-
What illness, injury, disease, or condition is the device intended to prevent, diagnose, or treat?
-
What are the circumstances under which someone would use the device?
-
What is the target population, the anatomical sites where it will be used, and the duration it will be used for?
It’s fairly common for MedTech companies to enter a market with a narrow scope in terms of the indications for use of their device. If they want to get to market quickly and have good predicates for their device, they may decide to go to market with a single indication for use, and then expand its indications later on.
This is one of the reasons I mentioned you should be keeping up with the off-label use of your device. It’s not legal to market your device for off-label use, but if you’re consistently seeing physicians use your device in additional ways they deem to be safe and effective, then the first step is to have your sales team begin capturing this intel. Once you have more information about this off-label use, then you can begin investigating whether you wan to add that indication to your device.
What to consider before adding a new indication for use
Before adding new indications for use, however, it’s a good idea to consider whether you’ve fully tapped the total addressable market (TAM) for your current indications. The reason for this is that, as we’ll see in a moment, adding new indications for use to your device isn’t always a simple process. New indications can potentially change risk classification, and in some cases, adding a new indication could trigger the need for a PMA (FDA has a useful guidance document on this topic).
Changing an indication for use may also change what you need from a device’s parts and components. If a new indication for use does move your device to a higher risk classification, you may need to evaluate whether your current parts are still suitable for the device.
For instance, maybe your device uses an aluminum screw with an anodized surface. You get some reports from users that the anodization on the device is worn through. If your device is low-risk and doesn’t make contact with any tissue, this may not be a big issue. But if you’re planning on adding a new indication for use, you need to know whether that will create a biocompatibility problem. You may then need a new screw or a new supplier.
If the addition of a new indication for use is going to bump you up a class, then you’ll need clinical data to support that new intended use; you may also need to look at a different predicate device that has all the indications for use you’re after.
And definitely don’t forget about reimbursement. Consider your current reimbursement strategy carefully and get clarity on how it may be affected by the changes to your indications for use that you’re considering.
These are the kinds of intricacies you can run into when you add a new indication for use to a device, especially if it increases the risk associated with the device. Of course, this isn’t to say you shouldn’t add new indications, just that it typically makes sense to tap into as much of your current market as you possibly can before going through the process of adding new indications.
2. Adding new products to your portfolio
For an established MedTech company, the majority of growth and expansion will likely come from adding new products to an existing product portfolio.
When we talk about adding a new product to a portfolio or product family, it’s essential that the intended use of that new device isn’t changing. If it does change, that will require the regulatory action we just went over in the above section.
Instead, a new product in a portfolio is often a different size or configuration of an existing device. In that case, it will essentially fit within your initial 510(k) and may not require any new submission. Again, though, carefully consider the FDA’s guidance or the relevant regulations in any other market you’re in before deciding you don’t need a new submission.
There are a number of considerations around adding a new device to a product portfolio, but the biggest one really centers around risk. Any time you’re adding something to a product, even if it’s just increasing its size, you must determine if that change creates additional risk. And if it does, you need to be able to prove you’re mitigating that risk.
Finally, don’t underestimate the cost and work involved in adding a product to your portfolio, as it could involve a lot more regulatory work and documentation than you initially thought.
3. Entering new markets
Another common expansion method is to start selling your device in new markets. And while this may seem like a no-brainer, expanding into a new market can hold some surprises if you don’t adequately prepare.
The regulatory side of expanding into any new market is critical and, as such, should receive a lot of attention. Understanding the new region’s regulations and your device’s relation to them will help ensure a smoother entry into the market.
But there’s also a business side to entering new markets that can’t be forgotten. Who are you selling to in this market? What are the market needs? What is the voice of the customer? What are the reimbursement options?
Sometimes MedTech companies make the mistake of marketing a device in a new market without understanding that market and what drives it. You should be able to clearly and comprehensively answer questions like:
-
Why are we doing this?
-
What’s the need that we’re meeting?
-
How are we going to meet it?
-
How will our device be sold in this market?
-
How do clinicians interact with us in regards to marketing and selling the product?
You may find that a new market has different players and differences in how devices are sold. Your reimbursement strategy will likely vary from one geographic region to the next, for instance.
![enterprise-guide-managing-medical-device-retiring-device]() End of the Line: Retiring a Device
End of the Line: Retiring a Device
 End of the Line: Retiring a Device
End of the Line: Retiring a DeviceYou may not be thinking about it now, but there will come a point when the device you’re managing (or perhaps just one of the products in your device portfolio) is ready for obsolescence.
Ensuring that the market is aware that this device is being discontinued is certainly important, but there are some other items to consider as well.
In fact, retirement is a lot like the initial development of the device, in that you need to go through a planning phase where you analyze the impacts of the device, both from a patient standpoint and economically speaking.
-
How much revenue does this device generate?
-
What is the cost of goods sold (COGS) associated with this device?
-
How much machine time does it use compared to other devices?
-
How long are you going to service equipment and when does that contract for service end?
-
Are you going to help the customer dispose of the device?
-
How and when will you be notifying them?
These questions highlight the importance of cross-departmental discussions when obsolescing a device. While a device might have a high return on investment (ROI) or sell in high volumes, it could also be monopolizing machine time that could be used for more expensive devices.
There’s also planning that has to happen from a regulatory standpoint. You’ll need to abide by any document retention policy that the relevant regulatory body in your market requires. This includes things like keeping batch records of the DHF, typically for the life of the device, plus two years. That means the expiry date of the last device to go to market, plus two more years.
Essentially, you need to make sure you can support this device to its expiration date and beyond. Devices with an expiration date should be disposed of by the customer, but some devices may not have such a clear cut expiration date. You may need to notify customers that this device is being discontinued, so that they can dispose of the device or return it.
The obsolescence of a device typically goes through a change order, just like any other change to the device. So, you would follow the same process outlined in 21 CFR Part 820.40, document control. You will need to describe the change, the justification for it, the people approving the change, the date, any supporting documentation, and what to do with devices that are currently being manufactured, devices that are being held in storage, and devices that are already in the field, and when those actions should take place. Will they be scrapped, used until depleted, or customers notified of discontinuance? It’s a business decision, and needs to be considered carefully.
![enterprise-guide-managing-medical-device-choosing-purpose-built-solutions]() From Design & Development to Obsolescence, Greenlight Guru Has Your Back
From Design & Development to Obsolescence, Greenlight Guru Has Your Back
 From Design & Development to Obsolescence, Greenlight Guru Has Your Back
From Design & Development to Obsolescence, Greenlight Guru Has Your BackKeeping a device on the market and making sure it continues to help improve the quality of life for the patients who need it is what everyone’s job boils down to in this industry. Getting a device to market is a huge win, but it means nothing if you can’t successfully manage that device to ensure its continued safety and effectiveness for end users.
At Greenlight Guru, we support MedTech companies throughout the entire medical device lifecycle. All our products are purpose-built for the MedTech industry, so whether you use Greenlight Guru for your eQMS or your eClinical platform, you can be sure that you’re getting software that is built specifically for you and your needs.
If you’re ready to see the power of our purpose-built software, then get your free demo of Greenlight Guru today!
Guide to Managing Your Medical Device on the Market & Scaling Manufacturing






