
Medical device startups have a pile of things to address yet often don’t have the pile of money to take care of everything that is needed.
Because of this, a medical device quality system often gets pushed to the back burner in favor of activities that are perceived to add more value and meaning to the startup.
Ignoring a quality system entirely until after development is complete and the product is launched is not a good idea.
A medical device startup should treat the development of a quality management system in a similar fashion as the development of a medical device.
Build your medical device quality system as you go. Bootstrap it!
Why bootstrapping works for a medical device quality system
If you have ever been an inventor, entrepreneur, part of a startup, etc., chances are you are familiar with “bootstrapping”.
The term bootstrapping has long been affiliated with startups. It often refers to self-funding, sweat equity — whatever it takes — to get to the next milestone.
Medical device startups bootstrap their product development all the time. The approach has merit for developing products.
Bootstrapping a medical device quality system makes good sense too.
Build your medical device quality system as you need it.
Understanding a medical device quality system
Keep in mind that a medical device company in the U.S. must comply with FDA 21 CFR Part 820 Quality System Regulations.
Other parts of the world also have defined regulations for medical device companies (i.e. EU medical device directives, Health Canada medical device regulations). And for the most part, if you have interest in markets outside of the U.S., your medical device quality system can be built based on ISO 13485.
Establishing a complete quality management system according to FDA and/or ISO can be a time-consuming process.
Time-consuming and often thought to add little value to the efforts of the medical device startup.
When you read and understand what a medical device quality system is all about, you quickly determine that over 75% of the FDA QSR and ISO 13485 requirements are addressing processes and procedures for post-product development.
Much of what a medical device quality system addresses pertains to production and post-production activities.
It’s no wonder that most medical device startups push a quality system until later.
Because of this, your medical device quality system can and should be bootstrapped.
QMS must-haves for early stage medical device companies
As noted, when you check out the quality system requirements, you’ll find that the vast majority is about production stuff.
However, there are parts of the quality system requirements that do pertain to medical device product development.
#1: Design control
In my opinion, having a solid defined design control procedure is the most important item to establish EARLY as part of your medical device quality system.
Why?
Design Control is a systematic framework for capturing key aspects of medical device product development to prove your product meets user needs and is safe and effective.
FDA defines Design Control regulations in 820.30 and ISO 13485 in section 7.3 Design & Development. FDA provides further explanation about what Design Control is in a guidance document (and it’s pretty good).
Fortunately, too, FDA and ISO are very much in alignment when it comes to Design Control / Design & Development.
I’ll break Design Control down into a short list of steps:
-
Capture User Needs (including intended use).
-
Establish a Design & Development Plan.
-
Define Design Inputs (all those requirements).
-
Have a Design Review.
-
Design your product. Document Design Outputs (drawings, specifications, etc.).
-
Have a Design Review.
-
Prove you designed the product correctly with Design Verification.
-
Have a Design Review.
-
Prove you have the right product with Design Validation.
-
Transfer from development to production.
Oh, and be sure to keep documentation and records throughout. Maintain these documents in a Design History File.
#2: Risk management
If design control is first, having a risk management procedure is a very close second.
Design control and risk management need to work hand in hand.
According to ISO 14971, the basic steps of Risk Management are shown in the infographic below.
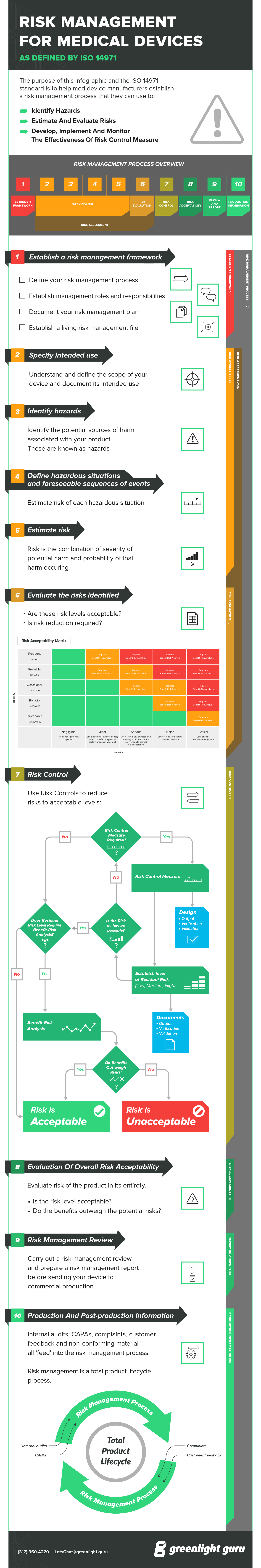
You can see how Risk Management and Design Control are complementary.
Keep in mind, too, that Risk Management is a HUGE focus of medical device regulators. Meaning the regulators expect you to establish Risk Management as a product lifecycle process.
You need to create documents and records throughout your medical device product development to demonstrate you have considered the risks and are doing something about it.
Align your Risk Management process with ISO 14971.
#3: Document control and records management
How do you create documents? Where are documents kept? Who approves them?
You probably noticed that I mentioned documentation and records under Design Control and Risk Management.
Guess what? Documentation and records are required throughout medical device product development and throughout manufacturing.
An old adage I heard many years ago is this: if it isn’t documented, then it didn’t happen.
Establishing a system for managing your documentation and records is critical. Not only during product development but throughout the existence of your company.
Ensure that you define how documents get approved, how you maintain revisions. Ensure you generate and maintain records resulting from product development.
Note, that the medical device regulatory world is still very much “old school”. We talk about document and records, usually because we visualize holding a pile of papers in our hands. There have been improvements in document control and records management. It is possible for you to maintain a system using digital formats.
#4: Supplier management
The reason supplier management makes this list is simple. Most medical device startups rely heavily on suppliers for many critical materials and processes.
I suspect your medical device product development depends on designers, material suppliers, test firms, etc.
You are buying materials and services from third party suppliers.
How do you know these suppliers are qualified?
When you qualify a supplier, you are doing so based on the specific material / service the supplier provides to you.
Your process needs to define how you qualify, evaluate, monitor, and manage your suppliers.
You need to keep an approved supplier list (ASL) too.
The rest of the medical device quality system is a waste of time (for now)
Yes, that’s right. I’m telling you to focus on what’s important to your medical device product development efforts.
Putting processes in place for Design Control, Risk Management, Document Control & Records Management, and Supplier Management and following them is plenty for your medical device quality system during development.
Of course, you will eventually need to put the rest of the QMS in place. And that can be done before you go to market and production.

BONUS RESOURCE:
4 Cornerstones of an Effective eQMS
4 Cornerstones of an Effective eQMS










