
Establishing a quality management system (QMS) is often seen as a burden—or at least a fairly high hurdle for medical device startups to get over.
The truth is that your QMS is a vital part of your organization’s success. And while it is a regulatory requirement, it may not be as difficult to build as you think.
In the US, the Food and Drug Administration (FDA) has published and makes available all of the regulations for medical device companies, including those for your quality management system (QMS). The QMS regulations can be found in 21 CFR Part 820, and cover everything you’ll need to do in order to build and maintain a compliant QMS.
For those outside the US (Europe, in particular), ISO 13485:2016 is the international standard for medical device quality management systems, and it can be purchased for a relatively small amount. The investment in the standard is absolutely worth it—and not only for companies outside the US. If you want a free resource to help along the way, check out our free Ultimate Guide to ISO 13485 here.
ISO 13485:2016 will soon be incorporated by reference into Part 820, creating what FDA has named the Quality Management System Regulation (QMSR). The QMSR will go into effect on Feb. 2, 2026, so it’s worthwhile to begin familiarizing yourself with ISO 13485:2016 now.
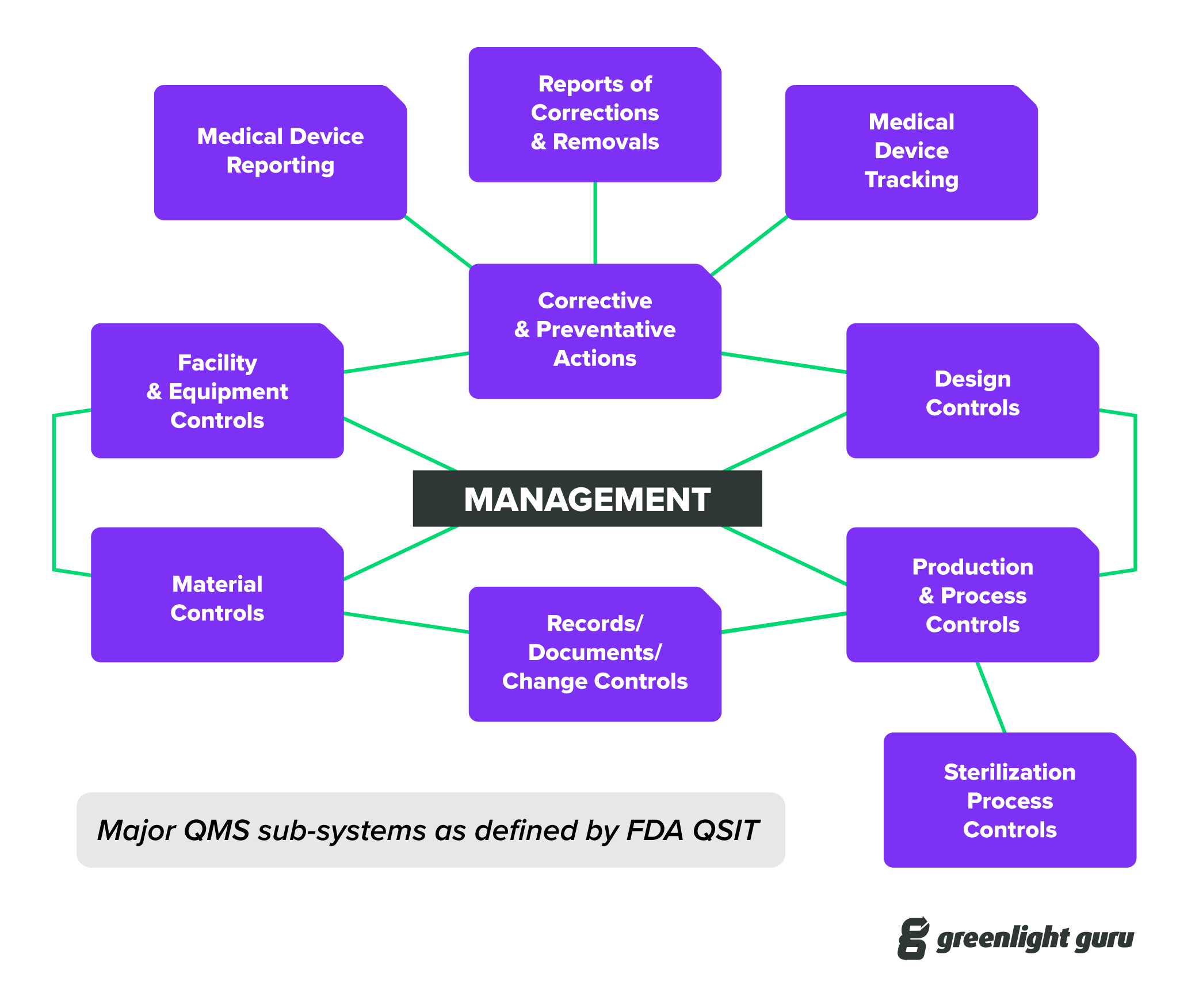
I’d also recommend taking a look at the FDA’s Guide to Inspections of Quality Systems, often referred to as QSIT. Getting familiar with QSIT is a bit like knowing all the questions on a test beforehand and having an answer key at your disposal.
For an ISO audit, you should review audit guidance documents available via International Medical Device Regulators Forum (IMDRF).
What I’m getting at here is that the requirements for your QMS have been clearly laid out, and there is a large body of guidance documents and information available to help you comply with those regulations.
How to implement a quality management system (QMS)
In this piece, I’ll guide you through the steps of building your QMS. Keep in mind that the order in which I suggest implementing your QMS is just that—a suggestion. You will need all the parts and pieces at some point in time in the genesis of your QMS. And the order may vary slightly depending on your product and company.
Regardless of whether your company has an established QMS or you’re just beginning your journey, this guide should help you as you consider where to start or what gaps you need to fill.
Let’s start with a couple pieces of advice that I think are pretty universal when it comes to building a QMS that adds value to your business and keeps you compliant without slowing you down.
Bootstrap your QMS
Every medical device company starts with an idea—and probably some funding. But not everyone gets funding, and not everyone gets the same amount of funding. Bootstrapping capital is a common tactic for startups, and you can use the same idea as you build your QMS.
With bootstrapping, you’re trying to get from one milestone to the next, adding value as you go. So, as you build your QMS, you should be thinking about what you need right now to get you to that next milestone, and what could wait a little longer.
For instance, if you’re in early stage product development, establishing QMS elements that are applicable to production and post-production may not be the best use of your time. Instead, focus your efforts on elements of your QMS, like design controls, that apply to the milestones you’re currently tackling.
Keep your QMS simple
It’s never a good idea to overcomplicate things when you’re just starting out. Your QMS needs to align with FDA and/or ISO 13485:2016 requirements. That probably sounds daunting, but remember, the regulations are telling you what you need to do, not necessarily how you need to do it.
In that sense, you have some leeway to create procedures that make sense for your company while still meeting the regulatory requirement. As you do so, keep three questions in mind:
- Does this meet the regulations?
- Is this as simple as possible?
- How does this impact the business?
The reason for the first question should be obvious—your procedures need to meet the regulatory requirements or you will find yourself in serious trouble during your first FDA inspection or ISO audit.
The reason for the second question is that when you’re starting out, overly burdensome or complicated procedures can slow you down tremendously without adding any real value. Right-sizing your QMS means meeting the regulatory requirements in a way that makes sense for the size of your organization.
The third question is a little trickier. I know that conventional wisdom suggests implementing a QMS may be somewhat disruptive to the business and may be viewed as not adding value. But I assure you that starting your QMS early and keeping it as simple as possible will add a significant amount of value to your company.
Having a QMS will help ensure that your company is generating the documentation and objective evidence that FDA inspectors and ISO auditors will expect to see.
With that in mind, let’s take a look at what you’ll need in your QMS during design and development, design transfer, go-to-market, and post-market.
QMS essentials during product development
If you’re bootstrapping your QMS and keeping things simple, you won’t need to have every single procedure in place during design and development.
But if you’re at the point where you have some funds and are pursuing design and development, you absolutely need to establish the first phase of your QMS, which should include:
- Design Controls
- Risk Management
- Document Control & Records Management
- Supplier Management
Design Controls
Design controls are a systematic framework for capturing key aspects of medical device product development to prove your product meets user needs and is safe and effective. You can find the framework for design controls in FDA 21 CFR 820.30 and in section 7.3 of ISO 13485:2016.
Risk Management
Risk management is a systematic application of management policies, procedures, and practices to the tasks of analyzing, evaluating, controlling, and monitoring risks related to your products.
The standard that we use for risk management in the medical device industry is ISO 14971:2019, but there are also plenty of references to risk management in Part 820 and ISO 13485:2016. In fact, regulatory bodies around the world expect you to establish risk management processes that align with ISO 14971:2019.
Document Control & Records Management
Requirements for document control and records management can be found throughout all of Part 820 and ISO 13485. Documentation is so important that one of the most common phrases you’ll hear in MedTech is, “if it wasn’t documented, then it didn’t happen.”
So establishing when documents and records are required and who needs to review and approve them is a crucial part of your QMS. While in product development, required documentation relates primarily to design controls and risk management.
But as your company evolves, document control and records management will continue to grow. Every step along the way will result in documents and records that will serve as the supporting evidence to prove you did what was expected. Establishing a sound methodology early, which can scale as your product gets to market, is essential.
Supplier Management
As a startup, chances are good that you rely heavily on outside suppliers for the products and services that you need to manufacture your medical device. Supplier management is about ensuring that you’re properly qualifying, evaluating, and monitoring those suppliers.
Remember, it’s not enough to assume that just because a supplier is registered with FDA or ISO certified that you can ignore your monitoring responsibilities. You still need to conduct due diligence to demonstrate that your suppliers are able to meet your needs and requirements.
And you still need records to demonstrate that you have implemented supplier controls commensurate with the criticality of the goods and services provided.
QMS essentials for transfer to manufacturing
To get your product from design and development and onto the market, at some point you will have to begin transitioning from development into manufacturing.
As far as design controls go, this generally starts to happen when you are entering Design Verification and Design Validation. Transferring to manufacturing is the time when prototypes and pilot production begins. This is the time when your product is about to be put through formal testing and analysis.
If you are conducting simulated use studies, animal studies, and/or clinical investigations, then your product should be transitioning to manufacturing prior to these events. When entering this phase, your QMS efforts also need to evolve to address these growing needs.
You’ll need to establish QMS procedures for:
- Training
- Purchasing
- Device Master Record
- Production & Process Controls
- Labeling & Packaging
- Receiving, Incoming, In-Process, Final Inspection
- Identification & Traceability / Device History Record
- Change Management
- Nonconforming Material
- CAPA
- Management Responsibility
Training Management
Training is a key process as your QMS evolves. You need to make sure that the right people are being trained on the right processes at the right time. To do so, you’ll need to put a training management procedure in place that identifies training requirements for personnel and provides a way for them to demonstrate proficiency with the skills they’ve been trained on.
Purchasing
Your purchasing procedures need to describe the minimum criteria required to buy goods and materials. Purchasing will go hand-in-hand with supplier management, as your goods and services should only be purchased from suppliers on your Approved Supplier List (ASL).
Device Master Record
Your Device Master Record (DMR) includes all the drawings, specifications, manufacturing instructions, etc. required to manufacture your medical device. Think of the DMR as the “recipe” for your medical device. This recipe is first established during product development because the design outputs you define during design controls are the preliminary DMR.
Production & Process Controls
Production and Process Controls are related to your DMR. You need to establish controls for your manufacturing processes in order to ensure reproducibility and repeatability. For some processes that you cannot verify completely via testing (like sterility), you will need to perform process validation.
Labeling & Packaging
You’ll also need to define what will appear on the labeling and packaging for your medical device, which also happens to be part of your DMR. Depending on your product and the risks involved in its use, the labeling and packaging specifications may be very important.
Receiving, Incoming, In-Process, Final Inspection
You will need to establish inspections and inspection criteria at a number of points throughout your receiving and manufacturing processes. These inspection points are meant to ensure the quality of your materials and components, as well as that of your finished device. And you must establish your specifications and acceptance criteria beforehand in order for the inspections to be meaningful.
Identification & Traceability / Device History Record
When you manufacture your devices, you must be able to establish identification and traceability of those products. Identification and traceability relates to the materials and components required for the device, often captured in a bill of materials (BOM), as well as your ability to know where products are, and in the case of a recall, your ability to retrieve product. The results of your identification and traceability are captured in a Device History Record (DHR).
Change Management
Changes to documents, records, goods, and materials are going to happen. Change management refers to the way that you manage those modifications to your products and processes in order to ensure that they are appropriately documented and implemented.
Nonconforming Material
Nonconforming material relates to any goods, materials, and products that fail to meet established specifications. Nonconforming material is also related to supplier management, as these nonconformances are often caught by inspections, either at receiving or later during the manufacturing process.
We recommend using a nonconformance report template that’s specific to medical devices. Every nonconformance may be a little different, but this template will help ensure that your nonconformance process is FDA and ISO-compliant.
Corrective Action and Preventive Action (CAPA)
Sometimes, issues with product or processes may rise above the level of a simple nonconformance. If a problem indicates a systemic issue or is severe in nature, then you will be required to open a formal investigation, known as Corrective Action and Preventive Action (CAPA). The goal of the CAPA is to get to the root cause of the problem, implement a solution, and verify that your action has solved the problem.
Management Responsibility
A key part of establishing your QMS is to ensure management has oversight and awareness. At least once per year, your company needs to conduct a management review to review all aspects of your quality system. You also need to name a management representative who will serve as the face of the company during FDA inspections and ISO audits.
QMS essentials for final go-to-market phase
Prior to bringing your device to market, you will need to establish and implement the final portions of your QMS. These procedures must be in place to ensure the quality of your product and capture feedback from users as it enters the market:
- Process Validation
- Software Validation
- Calibration
- Preventive Maintenance
- Handling, Storage, Distribution, & Installation
- Servicing
- Complaint Handling
- Adverse Event Reporting / MDR
- Corrections & Removals
- Customer Feedback
- Analysis of Data
- Quality Manual
Process Validation
Process validation is required for any processes where you are not able to verify the results 100%. It is also required in cases where you can verify 100% yet choose not to for business reasons.
Software Validation
Software validation is required for any software used in your company for managing aspects of your business impacting quality. This can include validation of QMS software, manufacturing inspection software, etc.
Calibration
Calibration relates to any gauges and equipment used to take measurements of product during manufacturing processes. The gauges shall be certified to recognized standards and updated periodically to ensure gauges continue to measure accurately and precisely.
Preventive Maintenance
Preventive maintenance applies to routine actions required to keep gauges and equipment operating as expected.
Handling, Storage, Distribution & Installation
You may need to define specifications regarding product handling, storage, and distribution. This can include temperature and humidity requirements. If your product requires installation at the point of use, you must also define the installation requirements and instructions.
Servicing
Servicing relates to any activities required to keep your product functioning and operational. This generally applies to reusable products rather than single-use devices. You’ll need to document all your servicing activities and keep records that are included in the DHR.
Complaint Handling
FDA defines a complaint as any written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution. You need to establish complaint handling procedures, including how you will investigate and address complaints.
Adverse Event Reporting / MDR
Any complaint about a serious injury or the potential for a serious injury related to one of your devices will need to be reported to FDA (and other regulatory bodies). For FDA, the mechanism for reporting is known as a Medical Device Report or MDR. Your procedures need to address adverse event reporting.
Corrections & Removals
Although you never plan to have a field correction or removal (otherwise known as “recall”), you have to establish procedures to deal with this possible scenario.
Customer Feedback
A complaint is a type of customer feedback. Complaints are generally reactive: you learn about the issue after it has occurred. However, ISO 13485:2016 requires you to establish customer feedback processes where you solicit feedback on the use of your products in a proactive fashion.
Analysis of Data
All of your QMS processes result in documentation and records. Ideally, you also establish key performance indicators (KPIs) and metrics to monitor your QMS performance. Analysis of data is one means to measure your QMS performance. Note that any data analysis must be done with proven statistical techniques.
Quality Manual
A quality manual is an overview of your QMS; it describes your company quality policy and provides brief descriptions of all the required quality system elements.
The purpose of a quality manual is to help you, your team, and any stakeholders or auditors navigate your QMS. Your quality manual should also communicate your company’s purpose and objectives for the QMS as well as establish the roles and responsibilities for maintaining the system and performing quality activities.
QMS essentials for post-market
Once your device is in the market and you have established your QMS, you need to define your internal auditing processes. You set the schedule and frequency for internal audits. It is important to make sure that personnel conducting internal audits have been appropriately trained to conduct audits. Oftentimes, internal audits are outsourced.
Internal auditing is a very important function. This is a way to monitor whether your company is following established procedures. Internal audits should be used as means for continuous improvements.
Greenlight Guru Quality gets your QMS up and running quickly, and scales with you
As I said before, keeping things simple—with the option to scale later on—is usually the best route for small companies with limited resources. And while many QMS systems tout their customizable and comprehensive software, they often leave out how difficult it is to implement and manage.
At Greenlight Guru, our QMS software is built specifically for medical device companies like yours. Because our software is aligned with 21 CFR Part 820, ISO 13485:2016, and ISO 14971:2019, you’ll have compliance built into your QMS from the start.
And with more than 70 customizable templates, you can get your QMS up and running quickly, focusing on the procedures that you need to have in place right now, while having the confidence that you can scale upward at any time.
Ready to see how a purpose-built QMS can help you right-size your QMS and accelerate your company’s growth? Then get your free demo of Greenlight Guru today →
FREE eBOOK: A Step-by-Step Guide to Complying with
ISO 13485 & FDA QSR
ISO 13485 & FDA QSR

%20QSR%20FDA-ISO.png?width=2550&name=(cover)%20QSR%20FDA-ISO.png)








