
It is my humble and expert opinion that the medical device industry has the most significant impact on improving the quality of life for all of humanity; more so than any other industry.
And I take this as my own personal responsibility to have a positive influence on helping to define and establish best practices, workflows, and processes to help other medical device professionals to design, develop, manufacture, and market the safest, most effective medical devices possible.
This is core to my being. And this is what led me to start Greenlight Guru, serving as the driving force behind the development of the only QMS software platform designed specifically and exclusively for the medical device industry by actual medical device experts.
The lifeblood of the medical device industry absolutely involves introduction of new and improved medical devices designed and developed to meet and address true needs, to improve and save human lives.

Why design reviews matter
The foundation of this lifeblood is medical device design controls. Design controls are a construct and series of activities and events that demonstrate, through documented objective evidence, that a medical device is safe, effective, and meets the needs of end-users and patients. Ultimately, design controls should unequivocally address two key questions.
-
Does the medical device demonstrate that the product has been designed correctly?
-
Does the medical device demonstrate that the correct product has been designed?
Defining design controls, while maintaining a design history file (DHF), throughout the design and development process are the key to addressing these questions. And throughout design and development, there are critical moments in time when it's necessary to check in and assess progress towards these ultimate objectives. These critical moments in time are design reviews.
Medical Device design reviews are events during design and development where certain aspects of product development are assessed for accuracy, appropriateness, safety, and efficacy.
Design reviews provide a systematic means to evaluate all design and development decisions. They provide an opportunity for feedback to product developers regarding potential and/or existing issues. Design reviews assess design and development progress, providing confirmation that design and development results are sufficient for progressing to the next stage of product development, and ultimately completeness of design and development.
Design and Development Methodologies
The term “medical device” covers a broad spectrum of products and technologies ranging from the classic, simple example of a tongue depressor, to electro-mechanical products, to implants, to single-use disposables, to software, to in-vitro diagnostics (IVD)...and the list goes on.
While there are guidance documents and standards that are specific to certain device types and categories, the core basic regulatory requirements are ostensibly the same. This is especially the case with respect to the design and development criteria applicable to medical devices.
With this in mind, how is it possible to build a quality management system (QMS) framework, including design and development workflows, to accommodate such a wide variation in product types?
When it comes to establishing a QMS and design and development framework for medical devices, a few key concepts come to mind:
-
Keep it as simple as possible
-
Establish agility and flexibility while ensuring compliance
-
Incorporate scalability
While considering the vast array of medical device types, these various concepts can present a challenge. And one area where I’ve seen companies struggle with simplicity, agility, flexibility, and scalability is with design and development best practices.
Having personally been a medical device QMS Guru and practitioner for over two decades, I have not seen many who have mastered this conundrum, even if their products are relatively similar in scope and complexity.
Let me just say this: Just because your medical device is complex does not mean that the systems and workflows that you use to capture and document design and development activities need to be equally as complex.
In fact, I would profess quite the opposite.
What you want from a design and development process is a system that provides “guardrails” to keep you compliant, while being nimble enough to accommodate the natural iterative nature of product development.
I’d like to debunk a common medical device design and development myth that I continue to hear all too often.
The Myth: To be compliant to regulations, medical device design and development must follow a phase-based “waterfall” methodology and approach.
Those who believe this myth are quick to point out the classic design controls waterfall diagram included within the FDA design control guidance document.

FDA Design Controls Waterfall Diagram
This is simply not true.
And FDA cited guidance corroborates this as a myth. (Realize that this guidance was published back in 1997 and is still applicable and appropriate today.)
“... In practice, feedback paths would be required between each phase of the process and previous phases, representing the iterative nature of product development. However, this detail has been omitted from the figure to make the influence of the design controls on the design process more distinct…”
And
“Although the waterfall model is a useful tool for introducing design controls, its usefulness in practice is limited. The model does apply to the development of some simpler devices. However, for more complex devices, a concurrent engineering model is more representative of the design processes in use in the industry.”
I realize that the term “concurrent engineering” may not be in vogue these days. In my opinion, this term is very similar to the current popular concept of agile development. Here are a couple of different definitions that expand on this term:
-
Concurrent engineering, also known as simultaneous engineering, is a method of designing and developing products, in which the different stages run simultaneously, rather than consecutively. It decreases product development time and also the time to market, leading to improved productivity and reduced costs.
-
Concurrent engineering (CE) is a work methodology emphasizing the parallelisation of tasks (i.e. performing tasks concurrently), which is sometimes called simultaneous engineering or integrated product development (IPD) using an integrated product team approach. It refers to an approach used in product development in which functions of design engineering, manufacturing engineering, and other functions are integrated to reduce the time required to bring a new product to market.
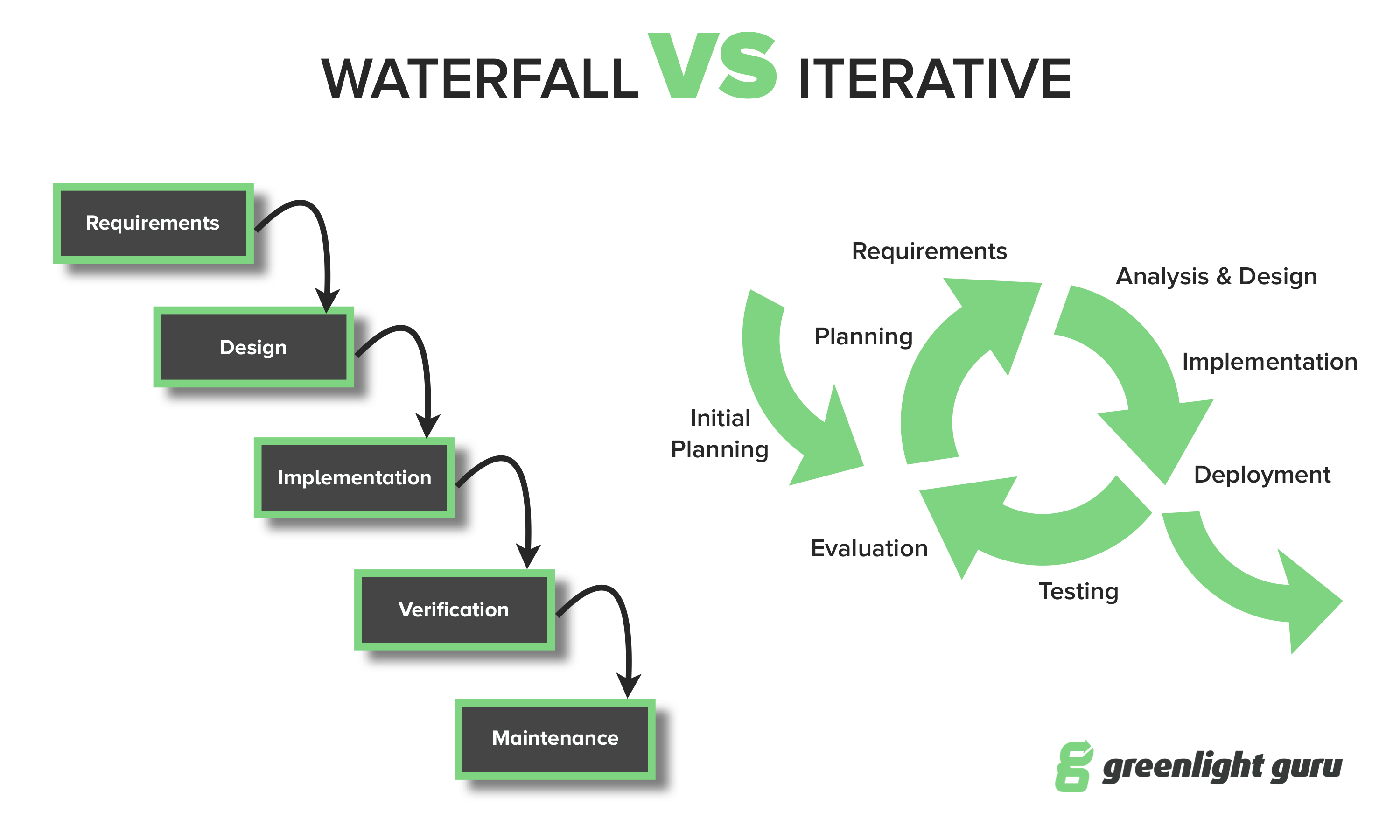
Regardless of your chosen product development methodology and the terms you use to describe it, this much is clear: design and development of medical devices can and should be an iterative process. And doing so is not counter to regulatory compliance.
I do want to spend the rest of this article providing some practical concepts regarding the simplification and flexibility of your design and development process. Specifically, I want to share some tips, pointers, and best practices when it comes to conducting design reviews throughout medical device product development.
What are Design Reviews?
Let me revisit the classic waterfall design control diagram.

Design Review Emphasis in Design Control Waterfall Diagram
Realize that this diagram is very important for medical device design and development to visually explain the relationships between design control elements. Again, to reiterate what I shared above, this diagram is not a prescriptive process for product development.
Notice the “review” box and arrows outlined in red that point to user needs, design input, etc. Why is this important and what is this implying?
Simply put, there is a need to conduct multiple design reviews throughout the entire design and development process.
How many? How frequent? Great questions and the answers are dependent on your specific device and internal practices.
Here are the specific requirements defined in ISO 13485:2016 and FDA 21 CFR Part 820 regarding design reviews.
ISO 13485:2016 Clause 7.3.5 Design and Development Review
At suitable stages, systematic reviews of design and development shall be performed in accordance with planned and documented arrangements to:
a) evaluate the ability of the results of design and development to meet requirements;
b) identify and propose necessary actions.
Participants in such reviews shall include representatives of functions concerned with the design and development stage being reviewed, as well as other specialist personnel.
Records of the results of the reviews and any necessary actions shall be maintained and include the identification of the design under review, the participants involved and the date of the review (see 4.2.5).
FDA 21 CFR Part 820.30(e) Design Review
Each manufacturer shall establish and maintain procedures to ensure that formal documented reviews of the design results are planned and conducted at appropriate stages of the device's design development.
The procedures shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed.
The results of a design review, including identification of the design, the date, and the individual(s) performing the review, shall be documented in the design history file (the DHF).
Design Reviews Should be Flexible
As the regulatory requirements indicate, design reviews shall be performed at “suitable stages” and “planned and conducted at appropriate stages.” This gives teams a great deal of flexibility on the scope and timing of medical device design and development review activities.
Note that is important to define the anticipated timing of design reviews as part of design and development planning efforts. And that planning, like actual product development, should be iterative and evolve throughout the process.
Another "must" aspect of design reviews to be aware of is that all design controls must be included as part of design review(s). Design reviews must also include appropriate functions and resources for the stage being reviewed and include an “independent reviewer.”
In order for you to embrace flexibility during medical device design and development, you need to define a product development approach that is also flexible. And your approach should align with the complexity of your devices and how your teams conduct product development.
Let me elaborate a bit on this concept of design and development process flexibility and how design reviews play into this.
Imagine you are developing a device that consists of a variety of sub-systems and major components. This could be an electro-mechanical device containing firmware and software components. This could be a software as a medical device (SaMD) product that has a number of modules. This could be a purely mechanical device that has connecting part and pieces.
Frankly, the type of device is less important. Having a construct that allows your product development team agility is more important.
Because with each type of medical device being designed and developed, there are different rates at which features and aspects will be defined and proven. There is a near constant state of iteration, especially earlier in the process.
Some product development teams and projects embrace more of a “divide and conquer” approach. This is especially true with electro-mechanical products where there are different sub-teams working on mechanical, electronics, firmware, and software aspects of the ultimate device.
And these sub-teams need the freedom to operate without employing cumbersome restrictions on the process. Each sub-team should have the ability to be as iterative and agile as needed without being so tightly tied and restricted by other sub-teams.
Yes, of course, there are moments in time during design and development when these sub-teams need to collaboratively work together. Which is exactly why there should be an emphasis on flexibility.
Each sub-team should be able to design and develop at a rate that makes sense for their particular scope while also aligning with the overall medical device, without being forced into a set of rules and details that constrict.
To break this concept down even further, the item or component being developed by the sub-team may yet still be broken down into additional “chunks” of work. This might be a bit too esoteric and conceptual. Let me be a bit more pragmatic to explain. To do so, let’s imagine that my sub-team is focused on developing a mechanical component of a particular device.
I will want to understand the form factor criteria. Where will this be used? Who will use it? Things like that. I may decide to make an initial proof of concept prototype of the form factor using foam or some other “quick and dirty” means. This prototype will help me better understand user needs.
I might also be able to define some specific design input requirements for the mechanical component. I probably have much more to learn and discover, thus am not ready to finalize all aspects of the mechanical component.
But there are aspects where I am confident and where it would be beneficial to formalize decisions about the mechanical component. And a key way to do so is via a design review.
Design Reviews in a Digital World
The aforementioned example is very common amongst product development teams, irrespective of the type of product or component.
But how are we, as medical device product developers, able to address this flexibility while also maintaining good documentation and staying in compliance?
In the increasingly connected and digital world we live in today, many medical device companies still resort to manually documenting design review activities in a disconnected, offline manner as a primary means to demonstrate compliance.
Seems a bit counterintuitive, right?
At least that would explain why product development teams across the board struggle with planning, conducting, and documenting design reviews as required by 21 CFR Part 820.30 and ISO 13485:2016. Especially for companies with highly technical products that require design reviews across mechanical, hardware, and software components.
This is a major obstacle that nearly every medical device company who designs and develops new products is faced with today. Their quality system used to document design controls, including design reviews, does not sync up with their real-time design and development process.

Enter Digital Design Reviews (DDR) from Greenlight Guru
On the heels of streamlining how medical device product development teams demonstrate traceability with multi-level design controls, Greenlight Guru is thrilled to further enable medical device product development teams with the launch of Digital Design Reviews (DDR).

Learn more about Greenlight Guru's Digital Design Reviews
Versatile enough to align with any existing product development methodology, DDR give teams the flexibility of including individual design controls or other components at different stages of development across distributed teams.
By eliminating cumbersome processes for documenting design reviews and bringing the design review process online, product development teams can leverage Greenlight Guru’s Part 11 compliant workflows and design review artifacts that are automatically included in a living Design History File (DHF).
Through the synchronization of design controls and design reviews, teams are equipped with a powerful solution that aligns with their actual product development process. DDR is a new approach included within the Multi-level Design Controls and Risk Management workflows in the Greenlight Guru eQMS software platform.
So, how does it work?
Recall the example described in the previous section which involved capturing various aspects of medical device design and development, while also being able to conduct flexible design reviews. Greenlight Guru DDR will enable you to do just that, all while ensuring documentation is maintained within your DHF and generating design review records, complete with full electronic review and signatures functionality.
At any point in time during a product development project, when you are ready to conduct a design review, selecting the specific items to include in a formal design review can be done in a matter of a few mouse clicks. And the beautiful part about DDR in Greenlight Guru is that your design reviews can be as simple or as complex as you need them to be.
With Digital Design Reviews, product development teams can effortlessly plan, conduct, and document their design review activities with actionable feedback. Teams can refocus their efforts on value added development activities that positively impact product submissions, launch timelines, and demonstrate regulatory compliance with confidence.
On a day to day basis, the team at Greenlight Guru has the opportunity to work with medical device companies all over the world who emphasize a culture of quality as a strategic imperative.
Greenlight Guru enhances those efforts through its industry-specific software and elite customer service. Together, we accelerate the development of innovative new devices that drive better patient outcomes, while also positively impacting both the top and bottom line for our customers.

FREE eBOOK:
Why Flexible Design Reviews Matter for Medical Device Product Development
Why Flexible Design Reviews Matter for Medical Device Product Development






