
How prepared is your quality management system for the new requirements of EU MDR for medical devices that will go into effect in less than a year?
The European Medical Device Regulation (EU MDR) has been created to replace the former Medical Device Directive (MDD) over a 3-year transition period, which comes to an end on May 26, 2020. These new requirements involve several changes that medical device companies must be prepared for, including things like device classification and updating your QMS. And most importantly, the EU MDR has formalized the expectations that your QMS, documents, records, product information, risk, etc. are all interconnected together. Said another way, all of the documentation and records of your QMS and products need to be a seamless system of data and information.
As you assess and prepare for EU MDR (and all of the other global medical device regulation changes), more and more emphasis will be placed on your QMS. Greenlight Guru's eQMS Software is exclusively focused on the medical device industry, and my top responsibility to you is to ensure that this solution keeps you ahead of these changes.
Regardless of the tools and solutions you use for your QMS, there are changes in EU MDR that have a direct impact on your QMS. Here’s what you need to know about preparing your QMS for these changes:

Overview of EU MDR
One of the first things to know about the MDR is that the regulation is more than four times longer than the old MDD. There is an increased emphasis on risk and safety, and a considerably expanded scope for “regulated devices.” Medical purpose devices and active implantable medical devices (AIMD) are now included in the MDR.
Previously, certain devices and accessories were not within the scope of the regulation. For companies, this means that you will need to conduct an assessment of your product portfolios and find where you may be impacted by the changes. You may have products on the market that were previously out of scope but now fall under MDR.
There are no more “essential requirements” as per MDD; under MDR, these are now “safety and performance requirements.” Along with these are increased requirements for clinical data. Previously a literature review or similar may have been sufficient, but some companies will find they now need data from clinical studies.
Another big change is that the deadline for reporting incidents that did not result in death or serious impact upon health has been changed from 30 days to 15 days.
I highly recommend that you begin working with your Notified Body now in order to figure out what applies to you and how your devices will be impacted. It is up to you to know the timelines for your devices and what you will need to do to ensure they are compliant with MDR.
Here is a key point to note: even though your device may be eligible for a grandfathering period, all QMS must meet the MDR regulations by May 26, 2020. Your audits after this date will be to the MDR standard, so it’s very important to be preparing your QMS now. These important dates are highlighted in red in the below graphic:

Overview Of QMS Impact For MDR
The EU MDR does have an impact on your QMS. While being certified to ISO 13485:2016 is important, this does not ensure that your QMS will be current with the new MDR criteria. EU MDR introduces some additional QMS expectations with respect to:
- Post-market Surveillance System
- Periodic Safety Update Report (PSUR)
- Incidents and Field Safety Corrective Actions (FSCA)
- Resource Management / Supply Chain
- UDI and Labeling
- Regulatory Compliance
- Document Storage Retention
- General Safety and Performance Requirements
- Implantable Devices
- Clinical Evidence
- Economic Operators
- EUDAMED
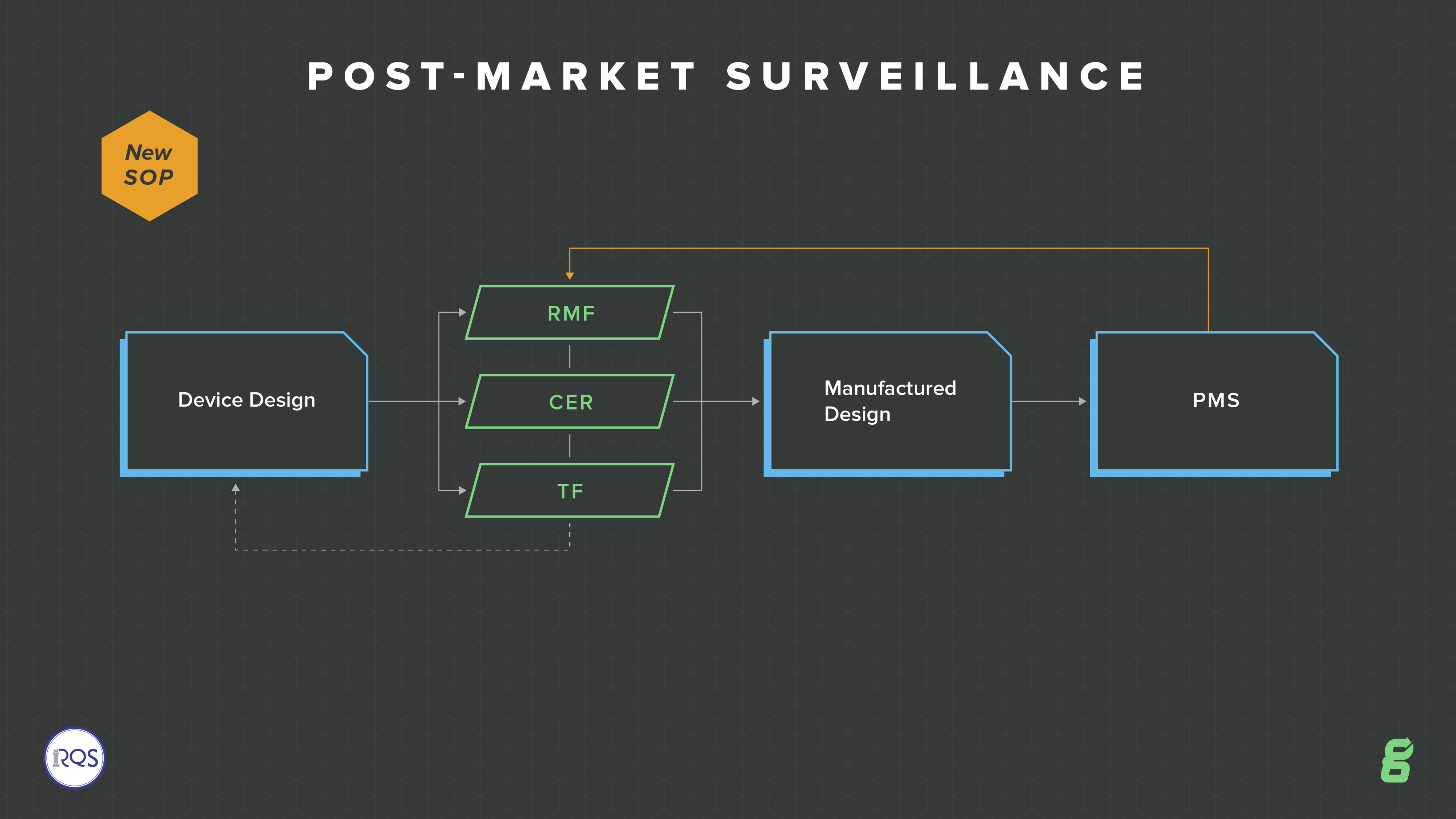
Post-market Surveillance
With increased requirements around compliance and vigilance, one of the big QMS changes is to post-market surveillance. The EU model wants to move more toward a whole lifecycle focus, so there is an emphasis on continuously updating your risk, your technical file and ongoing evaluation of your devices.
This expectation is so important for medical device companies to truly manage their devices throughout the entire product lifecycle--from design and development, through design transfer, through post-market surveillance, through product and process changes. How are you going to ensure all of these aspects are connected and tied together? Greenlight Guru's eQMS software is built out of the box to enable you to do just this.
You will need to have a post-market surveillance plan in place, along with procedures to satisfy the regulation. This means you will develop an ongoing feedback loop about your device, assessing whether risks or any other aspect need updating.
You are also required to produce a post-market surveillance report (PMSR). This is intended for low-risk Class I devices and it must summarize the results and conclusions of your Post Market Surveillance (PMS) data along with a rationale and description of any corrective actions taken for products on the market. This report becomes part of your technical documentation.
Periodic Safety Update Report
The Periodic Safety Update Report (PSUR) is addressed in Article 86 and is essentially an extension of the PMSR, but contains information for devices in higher risk categories. It covers classes IIa, IIb, III and implantables. It summarizes the results and conclusions from your PMS data. Additional information provides a summary of postmarket information, vigilance reporting, and current status of these devices on the market in the EU. You will also have to provide a rationale for any corrective actions.
This report must be updated annually for most devices, although Class IIa is only required to do so every two years. Additionally, any Class III devices or implantables must submit their PSUR to the EUDAMED database.
Incidents And Field Safety Corrective Actions (FSCA)
Incidents and field safety corrective actions (FSCA) is addressed in Articles 87 - 89. If you have a CE Mark currently, it is likely that you already have this covered in your procedures, but it’s worth checking to make sure you meet the new regulations. You need to ensure that your SOP outlines new requirements for reporting incidents and FSCAs.
Specifically, you need to ensure that:
- You detail timeline requirements for incidents and FSCAs
- Your SOP outlines statistical analysis of incidents and when to report to EUDAMED
- Your SOP incorporates risk into the investigation of the incident or FSCA
- You have templates or forms compliant with MDR.
Resource Management / Supply Chain
This section affects most medical device companies broadly. It’s not just about the company manufacturing the device, but it impacts distributors and importers too.
Article 25 represents a big change with a lot more information required to be documented and by whom. MDR identifies distributors, importers and EU authorized representatives as Economic Operators, each with specific responsibilities regarding verification of compliance, cooperation in complaint handling and field safety corrective actions, and cooperating with manufacturers and Competent Authorities in device traceability.
Your SOP must outline the process of device identification and traceability throughout the supply chain. Scrutiny over suppliers and outsourced processes is heightened. Each “economic operator” must confirm that the device is complying with regulations.
Some distributors and importers may never have had a quality system before. Under this regulation, they will need to develop one.
Tip: Be proactive about talking with your “economic operators,” ensuring that they are aware of the new regulations and are compliant.
UDI And Labeling
Those registered under the FDA will be familiar with UDI codes, but this is a new requirement under MDR addressed in Article 27.
You need to develop an SOP for UDI and labeling that:
- Defines storage of the UDI at each level (electronically is preferred).
- Details the process for importing UDI information into EUDAMED.
You are also required to keep an up-to-date list of all UDIs.
Note: At the time of writing this, no one knows how EUDAMED is going to work or what it will look like. It is still worth drafting a process for how you will get information to EUDAMED.
Regulatory Compliance
All companies should have within their quality system a defined strategy for regulatory compliance. Doing so might result in defining a new SOP.
You will discuss this in your management reviews, outline a plan, and have a designated person responsible for regulatory compliance. It should be documented who this is and what their roles are.
The EU MDR outlines the requirements of the “person responsible for regulatory compliance.” They must have a relevant degree or sufficient work experience to understand and meet the requirements of the standard. It also outlines that if you are a small company, you don’t have to have this person on-staff, but you should consult with a company that has the regulatory compliance expertise.

Document Storage Retention
The policy for document and storage retention isn’t new, but it has been updated. You must now retain the documentation for your devices for 10 - 15 years (depending on the type of device) after your last device was placed on the market.
You also need to be able to show that your QMS is effective throughout the extended storage period. This is one area where an electronic QMS such as Greenlight Guru will become an asset. In fact, Greenlight Guru is designed specifically for the medical device industry and has the necessary workflows and criteria in place to help you with transitioning to EU MDR.
It is also worth noting that you must have a plan in place to retain the documentation, should your company fail or go into bankruptcy. It’s unclear at this stage how this might be enforced, but nevertheless, it must be documented.
General Safety And Performance Requirements
One of the changes is that the previous MDD “essential requirements” are now referred to as General Safety and Performance Requirements (GSPRs). These must be included in your QMS and you must replace the reference to essential requirements with the GSPRs.
The high level of what is required is outlined in the diagram below. You then must have all of the templates and forms in place to meet the requirements.

There is a long list of items that are changing with the update to SPRs from ERs. It’s important to understand what those changes are by conducting a GAP analysis on your QMS. See below for a non-exhaustive list of some of the key requirements of MDR that must be fulfilled by the manufacturer:
- Risk management
- There is broadening of the scope of risk analysis, although typically, you may have already met this under ISO 13485:2016 and ISO 14971:2019. You need a risk management plan for each device, tighter risk analysis and risk controls, post-market risk procedures, and training on device safety as needed.
- Labeling
- There are additional requirements for labeling, such as the need to provide an SPR checklist. You are also required to provide information on warnings, precautions or contraindications on the labeling. For class III devices and implantables, a summary of safety and clinical performance is required on the labeling.
- EUDAMED
- You need a procedure in place detailing use and uploading of information to EUDAMED. You also need to include monitoring of EUDAMED. The system is set to go live in May 2022.
Implantable Devices
If you work with implantable devices, you will find that almost all of them are now Class III, as defined in Article 18. And with this, patient implant cards are required for all implantable medical devices which must be created for each device and reviewed under design control and risk.
All updates to the implant card should be available on your website as well as EUDAMED. Importantly, your SOP must document a plan for ongoing compliance with labeling and notification requirements.
Some additional points:
- Outside of implants, there are new requirements for devices that use human or animal tissue. You will need to comply with the GSPR section and the main text of the MDR on this.
- There are also new requirements for devices that use medicinal substances, for example, catheters that are coated with a drug.
- THIS IS OFTEN OVERLOOKED. You will need to update your Declaration of Conformity to state that you are now compliant with the MDR.
- The new program for harmonized standards and common specifications will be familiar to CE Mark holders but also has a role here. You must document how you will comply with and review the harmonized standards.
- There are many updates to IVDRs that you should review. For example, SOPs for companion diagnostics, product conformity and verification, and type examination.
Clinical Evidence
The importance of Clinical Evidence has been in place for a bit now with EU MDD. With EU MDR, there is even more emphasis being placed on clinical evidence.
While this may be obvious, the reasoning is about demonstrating continued device safety and intended clinical benefits and positive effects on patient health.
Clinical evidence is the combination of Clinical Data and Clinical Evaluation. Clinical data includes the safety or performance, clinical investigations, scientific literature for equivalent devices, and clinically relevant post-market surveillance information. Clinical evaluation includes continual analysis and assessment of clinical data and verification of safety, performance, and benefits.
Medical device manufacturers will have to ensure they have processes in place to ensure that there is sufficient quality data for their devices to conduct a qualified assessment regarding the product safety and intended clinical benefits have been achieved.
Also related to clinical evidence are changes to clinical investigation. Sponsors will need to obtain an identification number from EUDAMED and will require the use of electronic database for submission of applications and serious event reporting.
This will include a requirement for a clinical investigation plan, investigation brochure, and tie ins to verification and validation testing of the device.
Overall, the changes in clinical evidence put a much greater emphasis on product risk management throughout the entire lifecycle.
Economic Operators
EU MDR includes a new term called “economic operators”. Economic operators include manufacturers (Article 10), authorized representatives (Article 11 and 12), importers (Article 13), distributor (Article 14), and sterilizers and packaging (Article 22).
While manufacturers have always had responsibilities for medical devices, the requirements for the other economic operators are new and clearly defined within EU MDR with specific requirements that must be addressed.
EUDAMED
EUDAMED will be a database forum for manufacturers, distributors, authorized representatives, competent authorities, and the European Commission to exchange information about medical devices. Information such as product registration, declaration of conformity, economic operators, vigilance reports, and post-market surveillance.
At the time of this writing, there is a great amount of anticipation for EUDAMED, mainly because the database is still under development and not expected to be live until March 2020. And if you are following along so far, specifically as it relates to the timeline of EU MDR, the schedule for EUDAMED going live in time could present some challenges with several aspects of making the transition to MDR.
Summary - Preparing Your QMS
In summary, here are a few points to help you prepare your QMS for the EU MDR:
- Understand the key changes in the MDR and conduct a GAP analysis to understand where you are now, and where you need to be to comply.
- Compliance with ISO 13485:2016 QMS requirements will help you to comply with MDR too, as they are similar.
- You need a very practical system for risk management. Risk is a key focus in the changes. It needs to be focused across the entire lifecycle of the device.
- Ensure that everyone who is considered an “economic operator” for your device (suppliers, distributors, importers etc.) understand the changes and have a plan in place for compliance.
- You need a robust system for documenting your QMS. An eQMS is preferable, allowing you to easily keep it updated and transparent. Greenlight Guru's software platform is an excellent solution for this, including a built-in system for the technical documentation requirements.

FREE eBook:
Preparing Your QMS for EU MDR
Preparing Your QMS for EU MDR

%20Preparing%20Your%20QMS%20for%20EU%20MDR-eBook-Greenlight%20Guru.png?width=2550&name=(cover)%20Preparing%20Your%20QMS%20for%20EU%20MDR-eBook-Greenlight%20Guru.png)
%20Preparing%20Your%20QMS%20for%20EU%20MDR-eBook-Greenlight%20Guru.png?width=250&height=324&name=(cover)%20Preparing%20Your%20QMS%20for%20EU%20MDR-eBook-Greenlight%20Guru.png)



