
Throughout 2020, the medical device industry will see significant changes take flight. Among the biggest of these changes will be Europe's new medical device regulation (MDR) originally set to go into effect in May, although new uncertainties now exist around its timeline stemming from massive disruptions caused by the COVID-19 pandemic.
Regulations aside, the medical device industry is simply evolving from the outdated practices of the past. As you’ll see in this article, a growing number of industry experts and thought leaders are aware of the paradigm shift regarding technology, efficiency, and quality in the production and commercialization of medical devices. These changes are sure to have an impact on medical device companies of all sizes around the world.
To help companies get a better understanding of the industry's current landscape and what's to come, we asked five leading medical device experts to share their insights on the emerging industry trends in 2020, so you can position yourself and your company to make it your most successful year yet.
#1: Proactive Companies Will Be More Successful
Every medical device company should embrace and take a proactive approach to achieving true quality—here’s what one MedTech executive had to say about the importance of adopting a true quality mindset:

“In the EU I see many examples of regress to obsolete approaches to quality due to lack of time and resources because of the MDR. Some—those who I believe will survive—are adopting or working according to the true quality mindset. Due to new regulations and lack of guidelines, many are forced into more detailed analysis and strategies—which changes the approach somewhat from a mostly reactive mindset to a more proactive mindset.” - Claus Romer Andersen, Romer Consulting
This notion of a proactive approach to attaining true quality is fundamental to the vitality of any medical device company. Reactive companies wait until something goes wrong, then scramble to fix it, often in a rush. The stress and panic that ensue during those high pressure situations do not make for a productive working environment for teams, and, odds are, the issue will continue to resurface.
The way I look at it, if you’re waiting until it’s time to react to quality issues, you’re too late. A proactive approach allows you to allocate resources more efficiently and it also prevents most systemic errors from taking hold. Nearly every instance of a product recall—a manufacturer’s worst fear—could likely have been prevented by tackling quality head-on.
Here are two examples of true quality best practices that, when implemented right, are proven to be effective in helping companies succeed in their space.
Conduct Regular Design Reviews
There’s no exact number of design reviews that companies are required to conduct during the design and development process. While not carrying these out often enough can spell disaster, you can also overdo design reviews.
You may find yourself in a situation where you feel that not enough has changed to necessitate a review, and doing so would needlessly consume resources. Although that logic might make sense initially, reviews are, perhaps, the best way to step back and assess the state of your company and potential areas for improvements.
The U.S. Food and Drug Administration (FDA) recommends carrying out design reviews after each of the main product development stages, depicted in the waterfall model below:
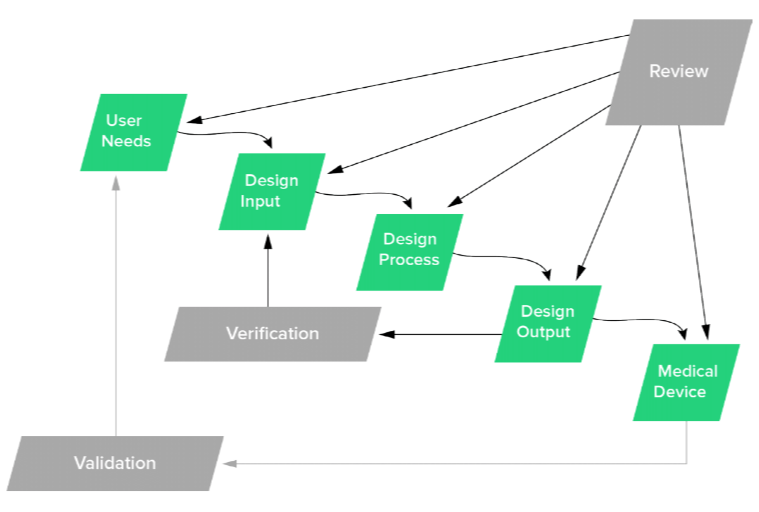
Published in the Design Control Guidance document, FDA states, "In practice, feedback paths would be required between each phase of the process and previous phases, representing the iterative nature of product development."
The purpose of structuring reviews this way is to ensure nothing is missed and that all aspects of your medical device are inline with where they should be through that point. As you can see, all elements of this process are intended to work in tandem with one another, which brings us to our next best practice around traceability.
Implement a Closed-Loop Quality System
A closed-loop quality system is one that connects the technology, people, and processes across every stage of the total product life cycle, from beginning to end. It’s much more difficult for systemic issues to take root using this kind of system that provides full transparency into all of your processes.
If and when issues do arise, a closed-loop system allows you to ascertain the root cause of issues so you can address them from the source. It can also provide visibility into other processes that may have also been impacted as a result, nipping ancillary issues in the bud.
Does that sound like your current method for approaching quality issues? If not, it may be time to consider implementing a closed-loop quality system that allows you to respond to issues in real-time and proactively monitor and mitigate risk. When it comes to ensuring the highest quality of your medical device, you need a quality system that will leave no stone left unturned.
#2: Executives Will Realize That Quality Adds Value
It's all too common to see manufacturers take a reactive approach toward quality. For these manufacturers, quality is an obstacle, not a goal. As you’ll see below, that mindset is evolving in the medical device industry:

“The industry is realizing that quality is value added, where previously it has been perceived as a burden and an unnecessary requirement. Due to the changing regulations, companies are beginning to see the value of quality.” - Adnan Ashfaq, Pharmi Med Ltd
Quality isn’t just about complying with regulations and bringing a medical device to market. Quality adds value to your company in many other ways as well—let's cover three ways in particular:
Reduced Regulatory Burden
Yes, as Adnan stated above, you need to focus on quality to achieve regulatory compliance. But focusing on true quality, quality for its own sake, will reduce the amount of effort needed for compliance, not make it harder.
If you’ve invested in quality early on, it's likely you'll face fewer problems during design verification, validation, and the launch of your product. That means fewer CAPA issues and fewer resources spent on lengthy internal audits and expensive changes to your medical device.
This also applies to managing your documents, a commonly cited pain point amongst manufacturers. Inefficient document management costs time and money. Investing in quality will pay off in the long run when it comes to managing documents and your design history file; the same is also true for other aspects of your day-to-day business, which you'll see become more streamlined.
Appeal to Hospital Systems
Historically, hospitals have been incentivized through receiving their compensation based on the quantity rather than quality of their services.
Value-based purchasing systems are becoming increasingly popular, however, with these systems holding hospitals more accountable for quality and value services offered. For example, the Centers for Medicare & Medicaid Services (CMS) organization rewards hospitals based on the quality of care they provide.
This renewed focus on quality may incentivize hospitals and service providers to partner with companies that share the same high-quality standards. Medical device companies that invest in proactive closed-loop quality systems will likely appear more valuable in the eyes of prospective partners.
Higher Valuations
In this case, the added value is quite literal. In my experience, VC firms are more likely to place a higher valuation on companies that are able to demonstrate a strong quality culture. Why?
Because those companies are more likely to produce safe, effective products. They’re more likely to navigate regulatory audits and inspections without any expensive delays. They’re more likely to have proactive systems and measures in place to detect minor issues that prevent major issues from happening in the future, such as product recalls.
Treating true quality as a goal sends the right signal to investors, and from what we can tell, more medical device executives are starting to realize this.
#3: Purpose-Built Technology Will Play a Bigger Role
The medical device industry has gotten by with paper and “digital paper” QMS solutions for decades. Paper systems are outdated, and the long-term costs of using a paper-based QMS can far exceed the original investment.
General-purpose QMS solutions, although an improvement in some ways, are not free from their own set of problems. These systems are cumbersome and difficult to configure to meet compliance and the unique needs of device makers.
Medical technology is evolving at a rapid pace—and this leaves little room for the outdated systems of the past:

“Changing regulations with ever-increasing requirements call for the implementation of efficient software-based quality management systems with the flexibility to adapt to the changing requirements. Fortunately, ISO 13485 has become THE standard to follow for quality management systems generally across the world, enabling one eQMS to cover all products and markets with higher efficiency.” - Brian Hedegaard, Probatus ApS
One thing that I believe every company needs is a single source of truth. If you’re taking paper documents from a binder, scanning them into a cloud storage system, and then collecting approval signatures via DocuSign®, how many sources of truth is that? How many single points of failure could exist in this system of manual processes?
Having the ability to create documents and procedures from pre-approved templates, collect digital signatures through Part 11 compliant workflows, and store and distribute those documents in a single electronic quality management system (eQMS) can catapult a company’s efforts for achieving success.
Leading medical device experts agree that eQMS solutions are indispensable tools for managing quality, and saving valuable time and resources:

“I see a shift towards software tools becoming more critical for QMS compliance and management. The use of the paper-based QMS will become the minority. New tools for collecting postmarket surveillance data and user communication will allow better communication between manufacturers and the end user.” - Kyle Rose, Rook Quality Systems
Companies will find that eQMS solutions will prove to be particularly useful for managing the large amounts of documentation, coordinating across teams that may be in remote locations, and staying prepared for whatever the outcome may be of our next trending topic of 2020, EU MDR.
#4: Biggest Challenge Will Be Uncertainties around EU MDR
2020 is here, which means the end of the three year transition period for EU MDR is upon us. According to recent news published by RAPS, however, the MDR deadline may be delayed by one year as a result of the disruptions caused by COVID-19.
Regardless of when the regulation goes into effect, transition efforts should still be prioritized by companies still not in compliance with the new regulation.
Among some of the biggest changes seen under EU MDR are:
- Increased requirements for clinical evaluation
- Digital and physical labeling
- Postmarket surveillance (PMS), and
- Total life cycle traceability
These stringent requirements raise concerns around whether companies will have the systems in place to adequately manage their documentation and quality:

“From a quality and regulatory perspective, the biggest challenge is MDR followed by MDSAP. Getting compliant with these regulations is the big challenge this year.” - Paul Rooney, Irish Quality Centre
The EU MDR will test companies in many ways. You may be faced with difficult decisions you must make around the financial feasibility of keeping existing products on the market due to newly incurred costs. You may find yourself scrambling to plug gaps in your quality system once the regulation officially goes into effect.
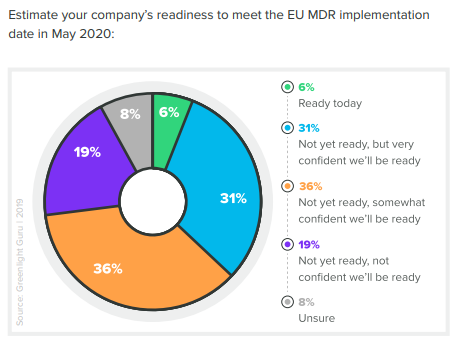
If you’re one of those companies, you’re not alone. According to our recent 2020 State of Medical Device Product Development and Quality Management Report, as of December 2019 only 6% of companies felt fully prepared for EU MDR.
The new regulation requires that postmarket reporting be handled within 15 days, at maximum—that is half of the previous limit outlined in the medical device directive (MDD), which is superseded by the MDR. Companies that may have taken their time in the past will need to place a new urgency in their processes to capture, report, and respond to complaints and customer feedback.
In addition, marketing materials, such as content published on manufacturers’ websites, will need to be kept up to date at all times and also outline all labeling information for the company’s devices sold within the EU marketplace.
Devices that were brought into the EU marketplace without a clinical evaluation must be tested in order to stay on the market, and be able to show full traceability of all processes within the product life cycle.
We’ve created a free EU MDR Gap Analysis Tool to help you find and assess the areas you need to update for compliance. When you’re conducting your gap analysis, consider the input from the industry leaders in this article. Their insights show strong indications that companies with reactive approaches or outdated methods will find it increasingly difficult to succeed in 2020 and beyond.
On the other hand, companies that embrace true quality and adopt a modern, proactive approach to developing and commercializing medical devices will position themselves for outlasting success.
Stay Prepared: Be Proactive
The best way to adapt to the changing tides of the medical device industry is to stay proactive. Take the time to assess the health of your company. Are your systems in working shape to manage the year ahead and into 2021? What possible points of failure can you find and begin working to fix in your company's culture or QMS?
The era of outdated quality systems is coming to a close. Put yourself and your company in a position of power by equipping your team with QMS software that prioritizes true quality, provides total product life cycle traceability while streamlining the processes throughout, and positions you for continued success in 2020 and every year after.
Looking for an all-in-one QMS solution to advance the success of your in-market devices and integrates your post-market activities with product development efforts? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
Free Flowchart for Improving your Quality Culture










