
Design controls are a set of quality practices and procedures that are used to ensure a finished device meets its user needs, intended use(s), and specified requirements.
The requirement for medical device companies to use design controls is enshrined in 21 CFR Part 820, as well as ISO 13485:2016.
The classic design control “waterfall” diagram looks like this:
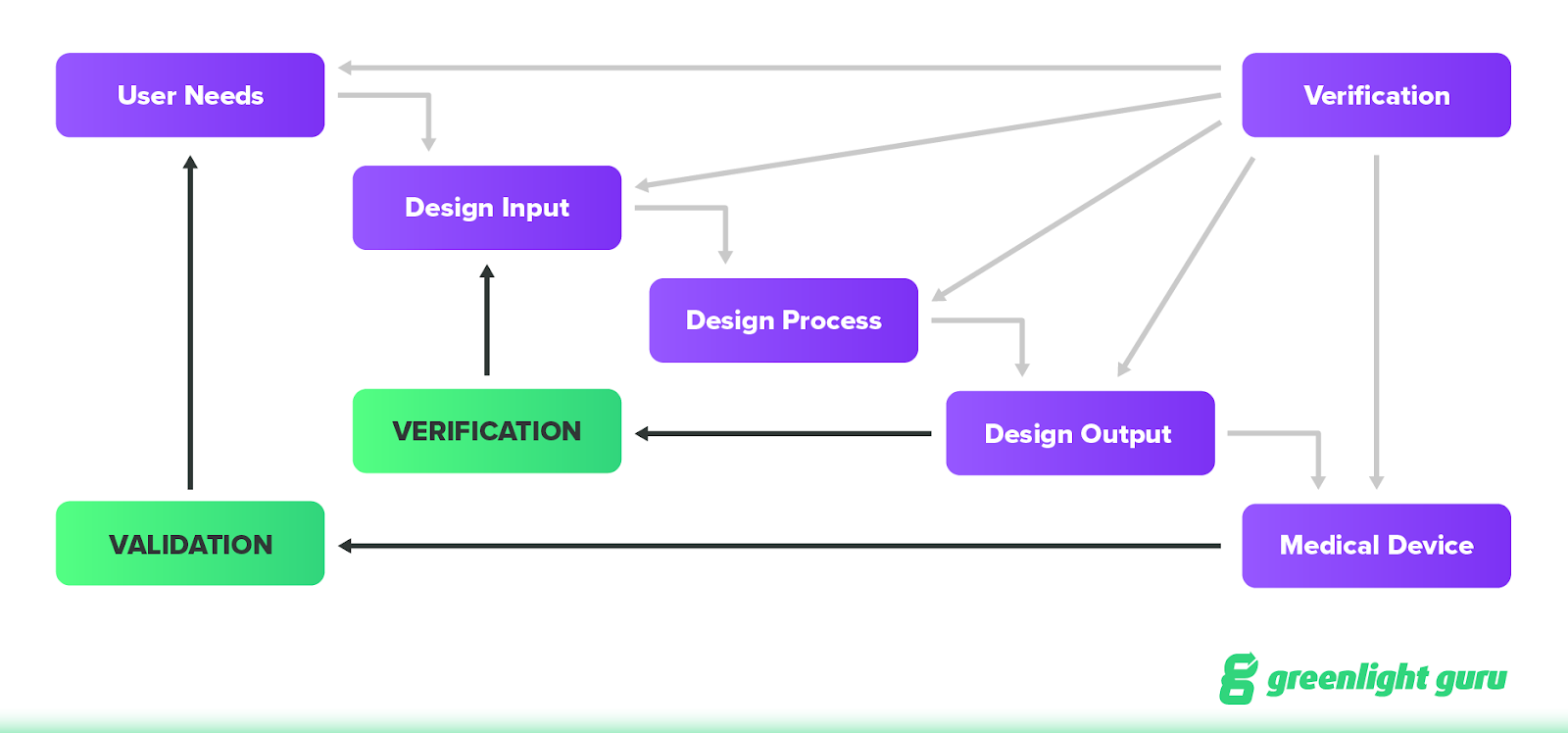
At a high level, the idea behind design controls is that they will not only improve the design of a device as the team goes through design and development, but also help to prevent future issues with a device once it’s placed on the market.
It’s important to remember, however, that design controls are not the only requirement specified by the 21 CFR Part 820. This regulation (and ISO 13485) describe the entire quality management system (QMS) requirements that every medical device company should have in place. Design controls play a central role in the QMS, but on their own, they’re insufficient to ensure safe and effective medical devices are being brought to market.
For the design control process to be truly effective, it needs to be connected to the other elements of your QMS so that your team can make decisions with the most up-to-date and accurate information possible.
Ideally, your entire QMS will be interconnected, but these are the five elements that you must be able to connect to design controls.
1. Risk Analysis
A risk analysis is our first critical element that must be integrated with your design controls. That’s because according to 21 CFR Part 820.30(g), design validation procedures must include risk analysis “where appropriate”.
The reality is, it’s almost always appropriate. Your risk analysis will identify potential hazards, including foreseeable misuse, and help you calculate the risks your device may pose under both normal and fault conditions.
Your risk analysis will also feed into your risk acceptability matrix, which will help you mitigate risk during the design and development process and ensure that changes to your design don’t introduce new hazards.
That’s why your risk analysis should be performed early in the design process. For example, once you’ve identified intended use, you should be able to identify cases of foreseeable misuse and off-label uses for the device.
2. Production and Processes Controls
21 CFR Part 820.70(a) directs manufacturers to develop, conduct, control, and monitor production processes to ensure that the device conforms to its specifications. That means establishing standard operating procedures, controlling your process parameters, and validating equipment and processes.
Think of it this way: if a piece of equipment is not operating properly or a test method you’re using to determine whether to release your product is faulty, then every decision you make about that product is now questionable.
Connecting your production and processes controls to your design controls will make life a lot easier for you if your company experiences a surge in demand and needs to increase manufacturing capacity. By connecting those production and process controls with design outputs and your risk analysis, you’ll be better able to maintain product quality while scaling.
For example, understanding critical-to-quality attributes and validating new processes can help manage the increased production without compromising quality.
3. Purchasing Controls
According to 21 CFR Part 820.50, manufacturers must also ensure that all purchased or received products and services conform to previously specified requirements. That means you need to have a plan in place for evaluating suppliers, contractors, and even consultants, and maintaining quality agreements with all of them. Effective purchasing controls are indispensable when it comes to maintaining the quality of components and services that you receive from third parties.
Your purchasing controls should connect to your design controls for the simple reason that your design controls specify the requirements for your device, and the components you receive from suppliers must meet those specified requirements in order to produce the device correctly.
4. Complaint Files
21 CFR 820.198 requires manufacturers to establish procedures for receiving, reviewing, and evaluating complaints, including adverse events involving their devices. An effective complaint management process can identify potential design issues with a device, which is why your complaint files should be connected to your design controls.
For example, if you were to experience a spike in product complaints or adverse events, an investigation might reveal a design flaw that could be corrected in order to prevent the same issues in the future. Or it may uncover that there is consistent user error of the device that was not considered in the risk analysis and may require new risk controls to eliminate.
5. Corrective and Preventive Action (CAPA)
In 21 CFR 820.100, you’ll find the requirement for corrective and preventive action, also known as CAPA, which includes processes for investigating the cause of nonconformities, implementing corrective or preventive actions, and verifying the effectiveness of those actions.
And while not every nonconformity should trigger a CAPA, connecting CAPA with design controls ensures that design-related issues are promptly addressed and prevented in future products.
Because CAPAs are opened when there is indication that a problem with a product or process is systemic in nature, it’s possible that an investigation will require changes to your product design, which is why CAPA and design controls should be connected in your QMS.
Greenlight Guru Quality is your fully connected, fully visible QMS solution
If the idea of integrating all of these QMS processes into your design controls feels overwhelming, then you may be using the wrong QMS solution.
At Greenlight Guru, our connected QMS software ensures that product development and quality processes like CAPA and Complaints are always connected, ensuring end-to-end traceability. And with fully integrated design controls and risk management, you can be sure you’re always working with the most up-to-date information.
Ready to see for yourself how a fully visible and traceable QMS can change the way you develop and manage your devices on the market? Then get your free demo of Greenlight Guru today →
Free eBook: The Ultimate Guide To Design Controls

%20Design%20Controls.png?width=2550&name=(cover)%20Design%20Controls.png)
%20Design%20Controls.png?width=250&height=324&name=(cover)%20Design%20Controls.png)



