
There are many global industries, but none quite like the world of medical devices. It’s not because we’re too niche or esoteric. It’s because no matter the device, the design, its applications, or where it’s from, every single one is united under the same mission of improving the quality of life.
But if you’re going to impact the world, sometimes those lofty goals require quite a bit of oversight. While some may grumble and groan at the mention of regulatory requirements, those interested in achieving True Quality know that these are in place to ensure public safety and the responsible manufacturing of medical devices. And perhaps nowhere is that better illustrated than with Unique Device Identification (UDI).
UDIs represent a game-changing way to accurately identify a medical device through the entire galaxy of MedTech in order to help patients, providers, and producers of medical devices. And while it’s easy to see UDI barcodes, product information reporting, and market-specific guidance as just another dose of bureaucracy, understanding what is being asked of manufacturers, as well as why these are in place is essential to success.
Let’s start by building our understanding of UDI from the ground up.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|












Table of Contents
| Choose a UDI solution purpose-built for your medical device to succeed |

![Ultimate guide to FDA Unique Device Identification System - What is UDI?]() What is UDI?
What is UDI?
UDI stands for Unique Device Identification, a distinct numeric or alphanumeric code used to identify medical devices throughout healthcare supply chains. These codes are generated by product owners based on globally accepted standards for device identification.
UDI codes can sometimes be overwhelming, especially if you’re new to the medical device industry. As a starting point, you should familiarize yourself with the two standard elements of a UDI code:
-
Device Identifier (DI) identifies the labeler and the model/version of a device. The DI is a required component and considered to be the static portion of the UDI, meaning it is the same for all instances of the product model/version.
-
Production Identifier (PI) identifies one or more variable characteristics, such as manufactured date, expiration date, lot number, or serial number. The PI is considered to be the dynamic portion of the UDI, meaning the PI value changes according to the production controls.
When combined, the device identifier and the production identifier form the full-length UDI. The DI is always first and is followed by the PI if it has a value. This is often shortened to the formula of UDI = DI + PI.
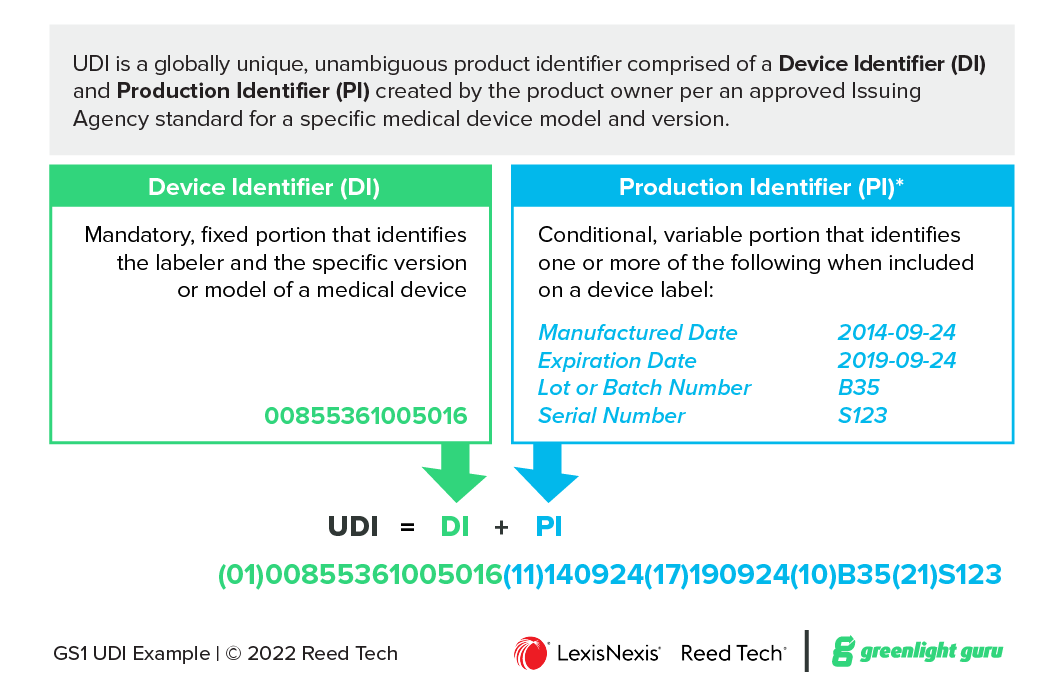 Figure 1 UDI Components
Figure 1 UDI Components
![Ultimate guide to FDA Unique Device Identification System - UDI Issuing Agencies]() Who are UDI Issuing Agencies?
Who are UDI Issuing Agencies?
Regional health authorities approve industry standards to define the format of the UDI code for their jurisdiction. The approved standards are managed by Standards Developing Organizations (SDO) and must meet various criteria for uniqueness, global scope, longevity, etc.
The following Issuing Agencies, also known as Issuing Entities, are commonly approved, but each health authority may create their own custom list:
-
GS1 — A nonprofit standards agency, GS1 sets international standards for supply chains, electronic data exchange, healthcare and more. GTIN (Global Trade Item Number) and UPC (Universal Product Code) are GS1 standards, both are numeric only codes. GS1 provides a “GS1 Company Prefix” to a subscribing organization and reserves a range of “Item Reference” numbers for the organization to assign to their product portfolio. GS1 charges an initial fee and a lower annual renewal fee for the subscription. GS1 is used by the majority of medical device manufacturers.
-
HIBCC – Health Industry Business Communications Council is a nonprofit organization focused on setting standards for the healthcare industry. The HIBC (Health Industry Bar Code) standard is managed by HIBCC and is an alphanumeric code. HIBCC provides a Labeler Identification Code (LIC) to an organization after paying a one-time subscription fee. The organization creates and assigns Product/Catalog Numbers (PCN) to their product portfolio without an additional charge.
-
ICCBBA – International Council for Commonality in Blood Banking Automation is a non-governmental organization. ICCBBA focuses exclusively on standards for medical products of human origin, such as blood products, tissues, and organs for transplant. It manages the ISBT 128 standard used in more than 75 countries. Only a few niche medical devices are identified using the ICCBBA standard.
Issuing Agency standards typically define a device identifier to encode a company, a product model, a package level, and a check character used to verify the accuracy of the code. Additional specifications provide guidance to create the production identifier portion of the UDI.
![Ultimate guide to FDA Unique Device Identification System - How does a UDI appear on a medical device?]() How does a UDI appear on a device?
How does a UDI appear on a device?
UDI codes are included on the product and package labels, and in some cases printed directly on the device itself. Regulations require the UDI be presented in both Automatic Identification and Data Capture (AIDC) and Human Readable Interpretation (HRI) format.
The AIDC format is typically a one- or two-dimensional barcode. This format allows the product to be identified swiftly and accurately by use of barcode reader and scanner technologies. The HRI text format refers to easily readable text which allows the product to be identified by humans without use of any scanning or barcode reader technology.
In some cases where the device is reused and reprocessed, such as a surgical scalpel, the UDI must be directly marked on the product, as the product packaging is discarded upon receipt. Various direct marking technologies are used, such as laser etching, to mark the product for the life of the product.
Issuing Agencies facilitate the reading of the UDI by adding additional alphanumeric codes to communicate the relevant information. These additional codes are called data delimiters.
Put simply, data delimiter codes signify the meaning and the format of the data that follows it. Think of these as flags within the UDI code, essentially letting the human or machine reader know that when they see a given data delimiter, the next alphanumeric string will contain a given data field. There are data delimiter codes for a wide array of information types, such as manufactured date, expiration date, lot/batch, and serial number.
The following examples illustrate the UDI presentation using the GS1 standard:
Device Identifier (GTIN): 00855361005016
DI with Application Identifier: (01)00855361005016
It should be noted that while some people interchange the GTIN with UDI, the GTIN identifier is GS1 Issuing Agency-specific and does not include the PI.
If the following production characteristics are present on the label, they must be included in the PI portion of the full UDI:
|
Label Symbol |
Label Text |
Label Value |
Corresponding GS1 AI |
|
|
Manuf. Date |
2014-09-24 |
(11) |
|
|
Expiry Date |
2019-09-24 |
(17) |
|
|
Lot No. |
B35 |
(10) |
|
|
Serial No. |
S123 |
(21) |
Table 1- UDI Symbols and Text
The full UDI with GS1 Application Identifiers (HRI text presentation) appears as:
(01)00855361005016(11)140924(17)190924(10)B35(21)S123
The full UDI with GS1 128 barcode (one-dimensional barcode) appears as:

The full UDI with GS1 DataMatrix barcode (two-dimensional barcode) appears as:

Where are UDIs published?
Once a UDI has been established, the data must be securely stored in the manufacturer’s repository and submitted to the respective health authority’s Unique Device Identification Database (UDID), such as with the US FDA Global Unique Device Identification Database (GUDID) or the European Database on Medical Devices (EUDAMED).
In general, regulations require product UDI data be reported to the UDID before the medical device is placed on the market. The UDI Device Identifier and numerous other data attributes, need to be collected and submitted to the UDID.
Product owners have some challenges in collecting these attributes as it typically is the first time this dataset is assembled. This is because the UDI data may be stored in different locations (siloed databases, file systems, local computers), may not be accurate or current, stored in varying formats, and have numerous data owners (some of which may have left the company).
Regional health authorities publish the submitted medical device information in their respective UDIDs and typically provide the content as a searchable, public-facing index for devices in a given market.
The database portals are available to any individual or organization, meaning all participants in the distribution supply chain can look up additional product information related to the device. The Device Identifier portion is the key in locating the correct record for a particular medical device.
Table 2- UDID Data
![Ultimate guide to FDA Unique Device Identification System - Benefits of UDI System]() What are the benefits of UDIs?
What are the benefits of UDIs?
An increasing number of health authorities are implementing UDI requirements to improve patient safety and the patient experience. Accurate, comprehensive product information gleaned from UDIDs is essential to ensure the correct and safe use of devices by all users, including patients, caregivers, healthcare providers, hospitals, and industry.
One of the primary goals of a UDI system is to ensure proper adverse event reporting among medical devices. Prior to UDIs, device identification was uncontrolled, allowing manufacturers to use their own various identification formats. This era made meaningful data analysis difficult, and as a result it was difficult to determine which specific devices were involved in a given incident.
UDI increases the accuracy of product traceability and enables medical device manufacturers, regulatory bodies, and other analysts to discover trends earlier and respond quicker in the event patient safety is at risk due to a certain device issue.
In addition to enriching adverse event reporting, UDI systems have been championed as a method to improve patient safety worldwide. The following three aspects of the UDI system establish a crucial foundation for the benefits provided by UDI.
Product Identification
-
Fewer medical errors caused by incorrect device identification
-
Better procurement, inventory, and administrative management in healthcare systems
-
Improved accuracy of product tracking and traceability throughout the supply chain
-
Better protection of the distribution supply chain against counterfeiting and diversion
Product Monitoring
-
Enhanced postmarket surveillance and effectiveness studies
-
Enriched adverse event reporting
-
More accurate analysis and earlier detection of safety issues
-
Improved development of corrective actions
-
Better management of recall campaigns
Product Information
-
Increased clinician, patient, and public access to device information via public UDIDs
-
More comprehensive digital health, electronic health records, and registries
-
Increased likelihood of using standardized, language-agnostic UDIs to link product information and adverse event databases across health authorities throughout the world resulting in improved patient safety.
![Ultimate guide to FDA Unique Device Identification System - History of UDI System]() The history of UDI: How did we get here?
The history of UDI: How did we get here?
The conceptualization of a new Unique Device Identification System established its roots back in 2007, when the idea for a global and singular tracking system piqued the interest of legislators in the United States, who instructed the FDA to develop and oversee new regulations to reduce medical errors, and allow defective medical devices to be detected and recalled earlier.
Over the next five years, FDA began running several pilot programs centered around the idea of a national, all encompassing tracking system. The first involved California’s Medicaid program (titled Medi-Cal, a delightful pun). The state agreed to implement a UDI requirement for device manufacturers. In this pilot program, FDA instructed state legislators to require the provider to submit a universal product number as part of the process for submitting reimbursement claims.
Over the course of the next two years, Medi-Cal data reported $30 million dollars saved in supply contracting. They also showed figures that the defective products in the state market were identified far more rapidly, meaning they were able to strike them from the list of covered Medi-Cal benefits. As a result, claims were more easily processed as the required information (such as the manufacturer's name) was rapidly accessible.
The second pilot program focused on the benefits for tracking device inventory at Mercy, a health system that operates hospitals and clinics across Wisconsin and Illinois, The UDI system was specifically implemented to monitor a coronary artery stent. The organization excitedly reported huge boosts in efficiency for their entire supply chain.
Because they had a new UDI-esque system, they were able to more quickly discover that there were huge oversupplies of inventory. Unique Device Identification also provided physicians with more accurate information on implanted medical devices and prevented procedure delays.
Following the huge success of these pilot programs and intensive preparation, FDA responded to the 2007 mandate from Congress and published a proposed UDI Rule on July 10, 2012. Roughly a year later after evaluating industry comments and revising the UDI approach, the FDA published the final UDI Rule in 2013.
Global Guidelines
In this same timeframe, the International Medical Device Regulators Forum (IMDRF) was actively authoring UDI guidelines. The IMDRF current membership includes Australia, Brazil, Canada, China, European Union, Japan, Russia, Singapore, South Korea, United Kingdom, and the United States of America.
This group prior to October 2011 was organized as the Global Harmonization Task Force (GHTF) and focuses on globally aligning medical device regulations on numerous topics.
The GHTF released an early UDI guidance, Unique Device Identification (UDI) System for Medical Devices in 2011 that established a global framework for local health authorities to conform to in order to harmonize UDI approaches across the world.
The IMDRF made some revisions and in 2013 released an updated guidance document, UDI Guidance: Unique Device Identification (UDI) of Medical Devices, which set forth fundamental concepts of a globally harmonized UDI system.
In order for a UDI system of such magnitude to even exist, IMDRF states that there are seven concepts that must be present:
-
The UDI and UDI Carrier are based on standards.
-
Any UDI applied to a medical device anywhere in the world should be able to be used globally and to meet the UDI requirements of its regulatory authority.
-
National or local identification numbers should NOT be a substitute for UDI.
-
Regulatory authorities should not specify the procedure for modifying these UDI standards.
-
The UDID core elements should not be modified.
-
The UDID should use the Health Level Seven International (HL7) Structured Product Label (SPL) and web based interface for data submission.
-
Every medical device needs to be identified by a UDI, unless it is exempt.
With those needs in mind, IMDRF’s elements for a harmonized UDI system are:
-
The development of the UDI using globally accepted standards
-
The application of that UDI on the label
-
The submission of appropriate information to a UDID
Is there a globally harmonized UDI System?
UDIs have the potential to change the landscape of medical devices for the better. However, global industries are complicated, and as UDI began to spread across the globe, it also created a new and unique issue of broadly similar, yet slightly differing, UDI requirements.
Each time UDI was rolled out in a new market, local regulatory bodies borrowed heavily from the IMDRF guidelines and early implementations, e.g., US FDA, but ultimately began constructing UDI systems and databases which relied on their own market definitions and approach.
In general, most of the variances across global regulatory markets revolve around:
-
Placement of the UDI
-
Content required in the UDI
-
Nomenclature and definitions
-
UDID portals
-
Use of carrier
-
Exceptions and special-case devices
Although many global core elements are found in regional UDI implementations. These small differences create variances between UDI policies and each UDI database. While variances may seem like a minor issue, remember that UDIs are primarily about product identification and the collection and maintenance of data; it’s difficult to combine datasets with variances. Sadly, the dream of a single global UDI system has not yet been realized.
![Ultimate guide to FDA Unique Device Identification System - UDI requirements in USA]() UDI requirements in the United States
UDI requirements in the United States
 UDI requirements in the United States
UDI requirements in the United StatesFDA UDI regulations were released in the final UDI Rule on September 24, 2013 and captured in the Code of Federal Regulations (CFR) at 21 CFR 801 (Subpart B) UDI Labeling and 21 CFR 830 Unique Device Identification.
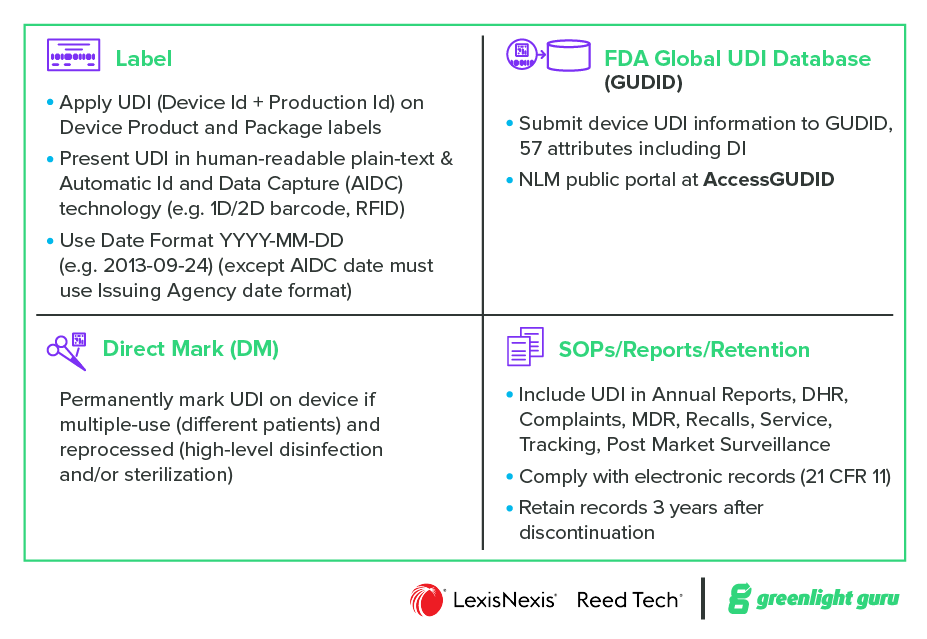
At a high level the US regulations establish the following requirements:
-
Label – Apply UDI (Device ID + Production ID) on Device Product and Package labels. Present UDI in human-readable plain-text & Automatic ID and Data Capture (AIDC) technology (e.g., 1D/2D barcode, RFID)
-
Direct Mark – Permanently mark UDI on the device if multiple-use (different patients) and reprocessed (high-level disinfection and/or sterilization)
-
GUDID – Submit device UDI information to GUDID, include 57 attributes including DI. (NLM public portal is at AccessGUDID)
-
Documentation (QMS) - Include UDI in Annual Reports, DHR, Complaints, MDR, Recalls, Service, Tracking, Post Market Surveillance. Comply with electronic records (21 CFR 11). Retain records 3 years after discontinuation.
The FDA UDI approach includes some noteworthy elements:
Labeler
The FDA created a term “Labeler” as the party responsible for UDI compliance.
…Any person who causes a label to be applied to a device, or who causes the label of a device to be modified, with the intent that the device will be commercially distributed without any subsequent replacement or modification of the label.
…in most instances, the labeler would be the device manufacturer, but the labeler may be a specification developer, a single-use device reprocessor, a convenience kit assembler, a repackager, or a relabeler
Issuing Agency
Each labeler is required to register with an FDA-accredited Issuing Agency to assign UDIs. FDA has approved GS1, HIBCC, and ICCBBA.
Timing
UDI Compliance Dates are phased by product risk class (subject to change):
2014-Sep-24 Class III Devices
2015-Sep-24 I/LS/LS Devices
2016-Sep-24 Class II Devices
2022-Sep-24 Class I Devices (label, direct mark)
2022-Dec-08 Class I Devices (GUDID)
Exceptions
-
Finished Inventory
-
Class I Good Manufacturing Practice (GMP)
-
Bulk Single-Use Devices of same version packaged together
-
Convenience Kits and Combination Products
Special Provisions
-
Standalone Software
-
Contact Lens
UDID
FDA created the GUDID, Global Unique Device Identification Database, to store submitted product UDI information. Labelers can submit product information to the GUDID via manual entry using the FDA GUDID Web Interface or electronically using XML file uploads via the Electronic Submissions Gateway (ESG).
The AccessGUDID database, hosted by the National Library of Medicine (NLM), was launched in 2015; it provides public access to FDA GUDID content and supports search, download, and web services.
UDI requirements in the EU![Ultimate guide to FDA Unique Device Identification System - UDI System requirements in EU and UK]()
European Union UDI regulations were released in 2017 and include the Medical Devices Regulation (MDR) 2017/745 and the In Vitro Diagnostics Regulation (IVDR) 2017/746.
These new regulations revamp the regulatory framework in the EU and incorporate UDI requirements. The European Commission (EC) provides a summary of EU UDI information at the UDI and UDI/Devices Registration websites.
 At a high level the EU UDI regulations have similar concepts as the US UDI approach, but there are numerous differences that must be understood and implemented.
At a high level the EU UDI regulations have similar concepts as the US UDI approach, but there are numerous differences that must be understood and implemented.
BUDI-DI vs UDI-DI
The EU UDI requirements initiated a novel EU product family grouping called Basic UDI-DI. Commonly referred to by its shorthand BUDI-DI, this product group identifier includes one or more related “child” medical devices, i.e., the UDI-DI which applies to specific devices. Manufacturers are responsible to create their own BUDI-DI identifiers per Issuing Entry standards and assign child device(s) to one group.
All BUDI-DI data attributes must be common for a product group, such as the same purpose, risk class, and various design or manufacturing characteristics. The BUDI-DI does not replace standard UDI-DI, nor is it used on any product labeling, physical marking, or AIDC data carrier. Instead, it is used for documentation, device registration, and identification in EUDAMED.
Company Identifier
The EC created a new company identifier, Single Registration Number (SRN), for identifying economic operators in the MDR/IVDR framework. Organizations can request a SRN by submitting an application to the ‘Actor’ module in the EUDAMED database.
Issuing Entity
Each manufacturer is required to register with an EC-accredited Issuing Entity to assign UDIs. EC has approved GS1, HIBCC, ICCBBA, and IFA.
Timing
Mandatory compliance dates for UDI/Device registration are dependent on the official EUDAMED launch date (subject to change):
2021-Oct-04 Start Voluntary UDI/Device Registration
2024-Q4 Start Mandatory UDI/Device Registration period
2026-Q2 Deadline Mandatory UDI/Device Registration period
Special Provisions
-
Standalone Software
-
Eyewear Products
UDID
The European Union Database on Medical Device (EUDAMED) is a digital platform for legal information on medical devices under the EU MDR and EU IVDR. Product UDI information, roughly 111 data attributes, is stored in the UDI/Device Registration module, one of six modules in EUDAMED.
Manufacturers can submit product information to the EUDAMED via manual entry using the EUDAMED Web Interface or electronically using XML file uploads via the Data Exchange access point. The reported product UDI product information is accessible via the EUDAMED database.
Medical Device Nomenclature
EU regulations require the use of a new standard named the EMDN (European Medical Device Nomenclature) in place of the commonly used GMDN (Global Medical Device Nomenclature).
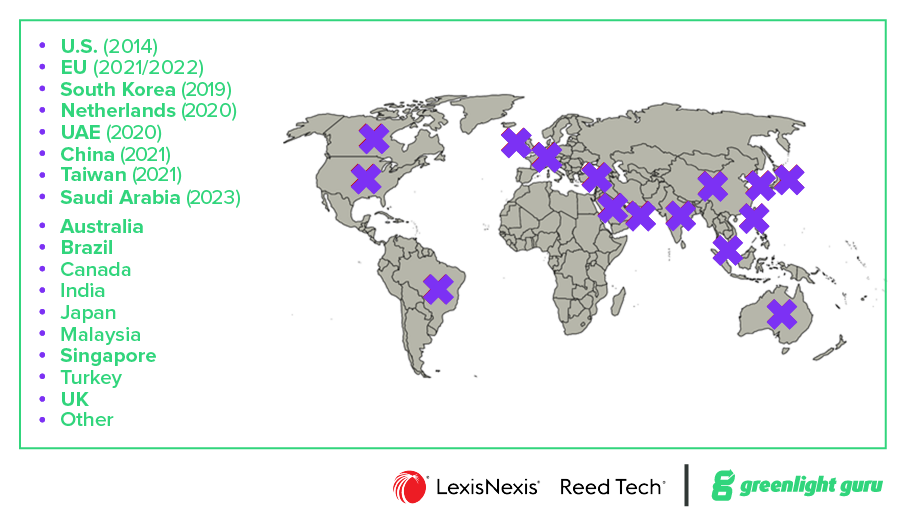
![Ultimate guide to FDA Unique Device Identification System - UDI Requirements Global]() UDI requirements in other regulatory markets
UDI requirements in other regulatory markets
In addition to the US and EU, numerous health authorities have or will in the near future require product information in UDI standards. For a deeper dive into these varied regulations, check out this exclusive Global UDI factsheet.
Tips on implementing a UDI solution for your medical device
For teams managing UDI regulatory data, the preparation, collection, submission, and maintenance details can quickly become a complex and burdensome task. The regulations are new, with varying levels of clarity and they are not harmonized. They have differing legislation, policy, timing, data, and submission approaches.
When implementing a UDI solution, whether it’s to comply with requirements from a single Health Authority or adding another region to your global UDI solution, it is critical to carefully plan your process to ensure a successful deployment.
Medical device UDI teams often find that engaging known experts in UDI database management helps ensure meeting deadlines and regulatory requirements.
Consider following the 5C’s when implementing a UDI solution:
-
Create a UDI Environment: As a foundation, assemble and train a multi-discipline team, create a customized plan for your product portfolio and deadlines, and update your systems.
-
Choose an Issuing Agency: Where you plan to market, and what financial model appeals best will help determine which Issuing Agency makes the most sense for you to choose. And while it’s always best to get it right the first time, don’t worry - it’s possible to change Issuing Agencies at a later time.
-
Collect Data – Build on your UDI solution; assign valid product identifiers, collect product attributes, enrich datasets and assign data owners/stewards.
-
Cleanse Data – Ensure the collected information is accurate, up-to-date, and complete. Implement data versioning and change management controls to govern multiple iterations of data cleansing. Allocate significant time and effort to produce high quality data records that pass data validation per business rules.
-
Comply With Requirements – Submit the product UDI records to the Health Authority repository. Apply UDI to product labels and begin production using UDI compliant procedures. Provide on-going maintenance to update/retire/add product UDI records and labels as necessary.
UDI implementation checklist and timeline
The following checklist identifies common steps in implementing a UDI system.

The time estimates for UDI implementation may vary considerably based on your existing level of UDI knowledge and compliance, the amount of effort and resources applied to the process and the number of products in your portfolio.
These estimates are based on the manufacturer engaging the existing Reed Tech global UDI solution Reed Tech SingleSource™ for Medical Devices, which provides a flexible, external data hub model with training support as needed.
![UDI and Software as a Medical Device]() How does UDI apply to SaMD?
How does UDI apply to SaMD?
As stated by the FDA UDI Final Rule (Section 801.50), Software as a Medical Device (SaMD) must comply with UDI requirements as other medical devices, but special UDI labeling provisions apply.
The other standard UDI requirements, submitting UDI product information to the GUDID and including UDI in supporting documentation/reports, are both fully applicable. Direct marking is not applicable to SaMD products.
Note that not all medical device software is considered “stand-alone software” and subject to the medical device UDI regulation. Many instances of software are included in a medical device as a component of the finished product and, as any other component, do not need to have UDI. In this scenario, only the finished medical device must meet UDI requirements.
All SaMD products must present the full UDI value (Device Identifier + Production Identifier) in an easily readable plain-text statement each time the SaMD is started, e.g., on the startup screen or flash screen, AND/OR in a menu command, e.g., on a Help/About page. The Automatic Identification and Data Collection (AIDC) format of the UDI, e.g., a barcode, is not required to be displayed upon startup or in the menu command.
Additional UDI labeling requirements differ depending on whether the SaMD is distributed in packaged form, i.e., physical media such as a CD-ROM, DVD, USB memory drive, or in a non-packaged form, e.g., an electronic download or cloud-based software.
For software distributed in a non-packaged form, the UDI presented as text upon startup and/or in the menu command must include the software version number as the “Lot/Batch” value in the Product Identification (PI) portion of the UDI.
For software distributed in a packaged form the UDI must also appear on the product and packaging labels in both easily readable plain-text and AIDC forms. Any Production Identifiers, e.g., Mfr Date, Expiry Date, Lot, or Serial Number, appearing on the label must also be embedded in the PI portion of the UDI. If a Version Number is on the label, it must be the “Lot/Batch” value in the PI portion of the UDI.
In the event the SaMD is distributed in both packaged and non-packaged forms, the packaged product DI and non-packaged product DI may be the same or different.
When do I need a new UDI?
As with most software, updates to SaMD products are fairly frequent. Sometimes, these are Minor Changes, such as fixing a coding error, patching a security vulnerability, or improvements to usability. In such cases, a minor change to SaMD does not require a new DI, as it can be identified with a new PI or Catalog Number.
When Major Changes occur, such as significant alterations that impact safety, performance, intended use, or effectiveness beyond the device’s original limits set by the manufacturer, the SaMD change must be identified as a new version or a model and therefore require a new DI.
Some examples of Major Changes to SaMD include:
-
Different algorithms
-
Database structures
-
Architecture
-
New user interfaces.
Should a major software change necessitate a new DI, manufacturers will also need to report the new DI and version number to the respective Health Authority as a new device record. Additionally, those new DI and versions need to be applied to the software and physical UDI label if applicable.
![UDI Solutions]() Choose a UDI solution purpose-built for your medical device to succeed
Choose a UDI solution purpose-built for your medical device to succeed
It’s sad, but they say all journeys in our world must eventually conclude. But as I said to you when we began, this world of medical devices is more than a little different. Standards and regulations are rarely set in stone; rather, they are in a constant state of evolution. And while it may be complex to keep track of moving targets, we know you didn’t get into this business because it was an easy road.
The UDI system has big changes on the horizon already, as EU regulation timelines continue to shift. In addition, many countries and markets around the world are only now beginning to require UDIs for medical devices. And to some extent, we know that many of you are at the outset of your journey as well.
So, in the spirit of both this guide and our MedTech world in general, we here at Greenlight Guru are proud to offer our QMS software purpose-built for MedTech.
Greenlight Guru is a Part 11-compliant solution that lets you seamlessly manage your electronic data in an end-to-end platform designed specifically for medical devices. We provide all the tools you need to take your device from design and development all the way through launch and postmarket. Even better—our world-class medical device Gurus will be with you every step of the way, delivering the expert advice you need, exactly when you need it.
Ready to take the next step? Contact us today for a free demo.
About the Authors

Gary Saner, Sr. Manager, Information Solutions Life Sciences, LexisNexis Reed Tech
Gary Saner is an experienced industry leader in establishing solutions for Life Sciences regulatory submissions. At Reed Tech, he is responsible for leading thought leadership and consultation for strategic planning and implementation of life sciences content management services including information capture, transformation, publishing, and lifecycle management.
He closely monitors regulatory regulations and activities, creates solution concepts for industry needs, and facilitates system development and implementation. Noteworthy projects include FDA Drug Labeling and Listing SPL submissions, EMA XEVMPD product information submissions, EMA/FDA Identification of Medicinal Products (IDMP), and FDA Medical Device Unique Device Identification (UDI) submissions.
Gary is actively involved with the HL7 SPL Working Groups: member of the SPL Leadership Team, SPL Process Team, and co-chair of the SPL Technical Team. He is a member of various industry groups: eStability, RPS, EMA SPOR Task Force, IRISS IDMP and industry associations: DIA and RAPS. He is a recognized authority within the life sciences community on SPL, FDA electronic Drug Registration and Listing (eDRL), and Medical Device UDI compliance. He serves on the Advisory Board of the LinkedIn Medical Devices Group.

Etienne Nichols, Medical Device Guru & Community Manager, Greenlight Guru
Etienne Nichols is a Medical Device Guru and Mechanical Engineer who loves learning and teaching how systems work together. He is also the Community Manager for the MedTech Excellence Community and hosts a weekly Ask Me Anything (AMA) or RoundTable discussion on relevant medical device topics within the community.
He has manufacturing, service, and product development experience, and has aided in the development of combination drug-delivery devices, and the UDI implementation for 250+ Class 2 medical devices. He has worked with both startup and Fortune 500 companies and holds a Project Management Professional (PMP) and Certified Scrum Master (CSM) certification. Having managed cross-functional teams to update designs of legacy products, Etienne understands the pains of a complex regulatory environment through the medical device development process.
His expansive knowledge, experience, and passion with medical devices is evenly matched with how much he enjoys helping customers work efficiently through the design and development process to bring safe, high quality products to market.
BONUS eBOOK: Ultimate Guide to UDI

%20Ultimate-Guide-to-UDI.png?width=1700&name=(cover)%20Ultimate-Guide-to-UDI.png)
%20Ultimate-Guide-to-UDI.png?width=250&name=(cover)%20Ultimate-Guide-to-UDI.png)



