The FDA has released a discussion guideline and request for feedback regarding changes in the certification process for medical device software that uses artificial intelligence and/or machine learning — see proposed regulatory framework here. This blog post explores what this FDA document means to companies who are involved in writing medical device software.
Artificial Intelligence in Software as a Medical Device
First, we should understand what is meant by Artificial Intelligence, or “AI”. This term is used frequently these days, often appearing in discussions of technologies such as autonomous vehicles, virtual assistants, and smartphones. A recent definition quoted in the FDA document defines AI as,
“the science and engineering of making intelligent machines, especially intelligent computer programs.” (McCarthy, J. – 2007 - What Is Artificial Intelligence? Stanford University, Stanford, CA.)
So, where legacy computer programs and algorithms operate with fixed instructions over a single set of input data to produce an output, intelligent programs and algorithms are more flexible and adaptable.
These intelligent programs typically draw upon additional data and algorithms that are external to the fixed instructions and input data and are capable of producing output that is more accurate than what is produced by legacy computer programs.
Machine Learning in Software as a Medical Device
A specific form of artificial intelligence that is of interest for use in Software as a Medical Device (“SaMD”) applications is the use of a generalization engine that can be trained to discriminate different inputs to predict an output. This type of algorithm that learns and improves the quality of output it produces is known as a Machine Learning algorithm.
More specifically, Machine Learning, or “ML”, is an artificial intelligence technique that can be used to design and train software algorithms to learn from and act on data. An algorithm so coded can use accumulated “training” data to dynamically adjust and modify its algorithm so that future calculations and output are more accurate.
For example, a ML algorithm could contain coefficients that affected the weights given to input statistics that affect the final outputs. Static or “locked” algorithms would produce exactly the same output for a given input set, and they are said to be Deterministic.
But a ML algorithm can adjust the coefficients with each run and store them for subsequent runs, and, therefore, its response to a given input set can change over time, producing a Non-Deterministic response.
One such ML algorithm is TensorFlow, a Python / C library with origins in the Google Brain project. There are other open- and closed-source frameworks available, with some targeted specifically at medical applications.
FDA Framework for AI/ML in Software as a Medical Device
With such recent developments in medical applications that utilize AI/ML techniques, the FDA is considering whether existing submission paths such as premarket clearance (510(k)), De Novo classification, or premarket approval adequately cover SaMD applications.
After all, typical submissions are of software and systems that are “locked” and are intended to be used as such, with the assumption that individual devices with the same production configuration will behave in the same manner as the approved device. However, AI/ML SaMD does not necessarily provide the same assurance.
Over time, as the devices are exposed to different data sets and accumulate more training and actual usage, their results can individually “drift” somewhat, leading to different internal conditions and potentially different responses to the same input data.
The FDA guideline presumes that software so developed may not conform to the existing certification process, such that adaptations and extensions to the certification process are needed. So, how does the FDA propose to extend the regulatory process?
First let’s look at the categories of modifications the FDA is examining. As defined in the guidelines, these are grouped under:
- Performance – improvements related to analytical and clinical performance, with no change to intended use or input type
- Inputs – changes in inputs with no change to intended use, and
- Intended Use – a change in the significance of the information provided by the SaMD
By including guidelines for the development and release environment and processes, the process seeks to ensure that subsequent releases conform to the original certifications, or that the certifications are revised appropriately, or that an additional review and certification process is triggered prior to release.
The FDA is presenting this as a Total Product Lifecycle (“TPLC”) Regulatory Approach that observes both the pre-market development to post-market performance along with examination of the “organization’s excellence.” (See Figure 1 below.)
 Figure 1 - Total Product Lifecycle Approach
Figure 1 - Total Product Lifecycle ApproachSo, how is the “organization’s excellence” reviewed and qualified? The FDA outlines a two-fold approach that seeks to:
- Assure the usage of Quality Systems and Good Machine Learning Practices (GMLP) by the organization, and
- Assure the usage of SaMD Pre-Specifications (SPS) describing the modifications and Algorithm Change Protocol (ACP) processes to achieve the changes and control the risks.
These approaches work together to determine the level of FDA review required for new modifications. As an example, a change that solely increases performance, is consistent with the SPS, utilizes existing ACP, and did not change intended use or inputs could be made without additional FDA review.
On the other hand, a change to Intended Use would require an FDA review of new SPS and ACP before the change is permitted.
Figure 2 below shows the proposed decision-making process for evaluating new releases of an SaMD.
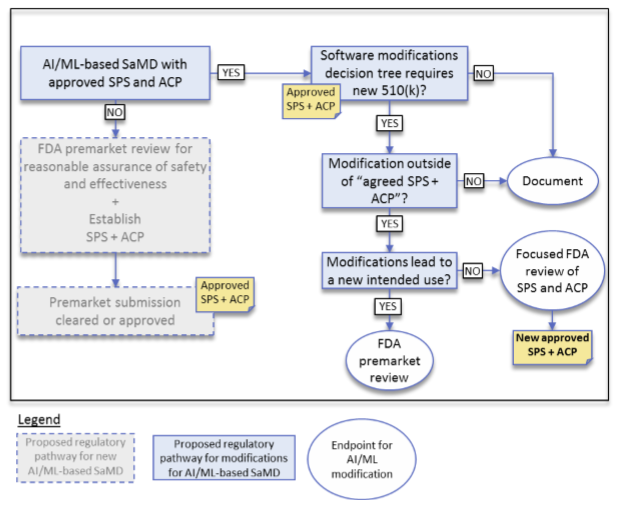
Figure 2 - Algorithm Change Protocol Components
Conclusion
While in a draft form, the FDA publication regarding the proposed regulatory framework for SaMD’s shows the complexities of dealing with AI and ML-based software and the serious thought the FDA has given thus far to supporting use of AI and ML in medical device software.
For any organizations contemplating use of AI or ML software, being aware of the regulatory framework proposal from the FDA can aid in planning for both successful launch and for on-going update releases of SaMD software.
More Information
-
US FDA Artificial Intelligence and Machine Learning Discussion Paper
-
Presentation by Finale Doshi-Velez from the Harvard School of Engineering and Applied Sciences
-
Challenges in the Verification of Reinforcement Learning Algorithms
This article was originally published by The RND Group and is being republished here with permission.
Looking for an all-in-one QMS solution to advance the success of your in-market devices that can integrate your post-market activities with product development efforts? Click here to take a quick tour of Greenlight Guru's Medical Device QMS software →
Greenlight Guru QMS Software