
The FDA’s medical device approval process is not the same for every device, and the timeline for regulatory approval can vary widely depending on the risk posed by the device. Given that tongue depressors, MRI machines, and everything in between are defined as medical devices, it makes sense that there is no quick and easy answer to the question, “How long will it take?”
However, depending on the regulatory pathway required for your device, there are general timelines that you can expect from submission to approval. In this article, we’ll walk through the most common regulatory pathways, their timelines, and the factors that determine them.
BONUS RESOURCE: Click here to download a free 510(k) submission checklist!
How long does it take to get FDA approval for medical devices?
Depending on the risk associated with your device, the medical device approval process can take anywhere from 90 to 180 days. However, it’s important to note that these numbers can be (and often are) increased if FDA requires a written response or additional information from the manufacturer. Some regulatory pathways, like Premarket Approval (PMA), can take far longer than 180 days, especially if there are deficiencies in the initial application.
Keep in mind, most class I and some class II devices are exempt from a premarket notification submission to FDA for approval. Most class II devices use the 510(k) pathway to market, which can take up to 90 days for FDA to review and respond. All class III medical devices must go through the Premarket Approval (PMA) process, which takes at least 180 days for FDA approval.
These are, however, general estimates, and there are nuances to each regulatory pathway that can affect the length of time the FDA's medical device approval process takes.
Four common pathways to market approval
The FDA’s approval process for medical devices varies by the regulatory pathway taken.
In this section, I’ll walk through four of the most common medical device approval pathways and link to some information on less common pathways at the end.
Self-registration
Most class I medical devices and some class II medical devices are exempt from the 510(k) requirements, meaning they do not require prior approval from FDA to market them. They are, however, still subject to general controls and other requirements such as establishment registration and device listing.
510(k) submissions
The 510(k) pathway is meant for class II medical devices that have not been deemed exempt from premarket notification. Section 510(k) of the FD&C Act requires manufacturers of these devices to notify FDA of their intent to market a medical device at least 90 days in advance of doing so. This premarket notification gives FDA time to determine whether the device is substantially equivalent to its predicate device (a device already on the market).
A 510(k) may be used for both:
-
New devices that a manufacturer intends to market for the first time
-
When a manufacturer intends to reintroduce a device that has been significantly changed or modified to an extent that might affect its safety or effectiveness.
The 510(k) process includes a comprehensive review of the safety and performance data for the device, potentially including scientific, non-clinical, and clinical data. FDA states that their goal is to make a decision on a 510(k) submission within 90 days after they receive it. However, this timeline excludes any “hold days” which can occur for a number of reasons, including an Additional Information Request during the substantive review.
Premarket Approval (PMA)
The Premarket Approval process is used for all class III devices, and it is the most rigorous regulatory pathway for medical devices. FDA’s review of a PMA submission is a four-step process that consists of:
-
Administrative and limited scientific review to determine completeness of the application
-
In-depth scientific, regulatory, and quality management system review
-
Panel review by an appropriate advisory committee
-
Final deliberations, documentation, and notification of the FDA’s decision
FDA will complete its review of the PMA application and advisory committee’s report and issue a decision within 180 days of the PMA filing. However, there are four types of decisions FDA may issue:
-
Approval order
-
Approvable letter
-
Not approvable letter
-
Order denying approval
What’s important to know here is that an approvable letter is not an approval. It will come with specific additional information or conditions that the manufacturer must meet in order to gain approval.
Similarly, a not approvable letter will describe deficiencies in the application and may identify what is necessary to make the PMA approvable. In both cases, the manufacturer has 180 days to respond in writing to the letter.
As I mentioned earlier, the initial 180-day timeline for the FDA’s medical device approval process can be misleading without context. Depending on the nuances of your device and application, it could easily take twice as long to gain approval.
De Novo classification request
A De Novo classification request is a less common regulatory pathway that medical device companies may use to get to market. Novel devices (meaning those that do not have a predicate device on the market) that are low-to-moderate risk may request De Novo classification from FDA.
The De Novo process also includes a substantive review, and if FDA identifies deficiencies, they may first ask for them to be addressed via an interactive review. If the issues and deficiencies can’t be addressed through an interactive review, then FDA will send the manufacturer an Additional Information request.
The FDA’s goal is to make a decision about De Novo classification requests within 150 review days (which exclude days the request was on hold for any reason, including Additional Information holds).
Tips for a smooth FDA approval process
Though there are many routes in the medical device approval process, there are a few steps that any MedTech company can take to ensure as smooth a path as possible. For anyone bringing a device to market, I’d recommend that you:
Start your regulatory planning early
FDA has stated that the average 510(k) submission is now more than 1,000 pages long. If you just did a double-take, you’re not alone. The 510(k) is technically the “easiest” route to market aside from self-registration for class I devices, and it’s still an enormous submission to put together.
Here’s a visual breakdown of the pathways (including the Medical Device User Fee Amendments) for comparison:
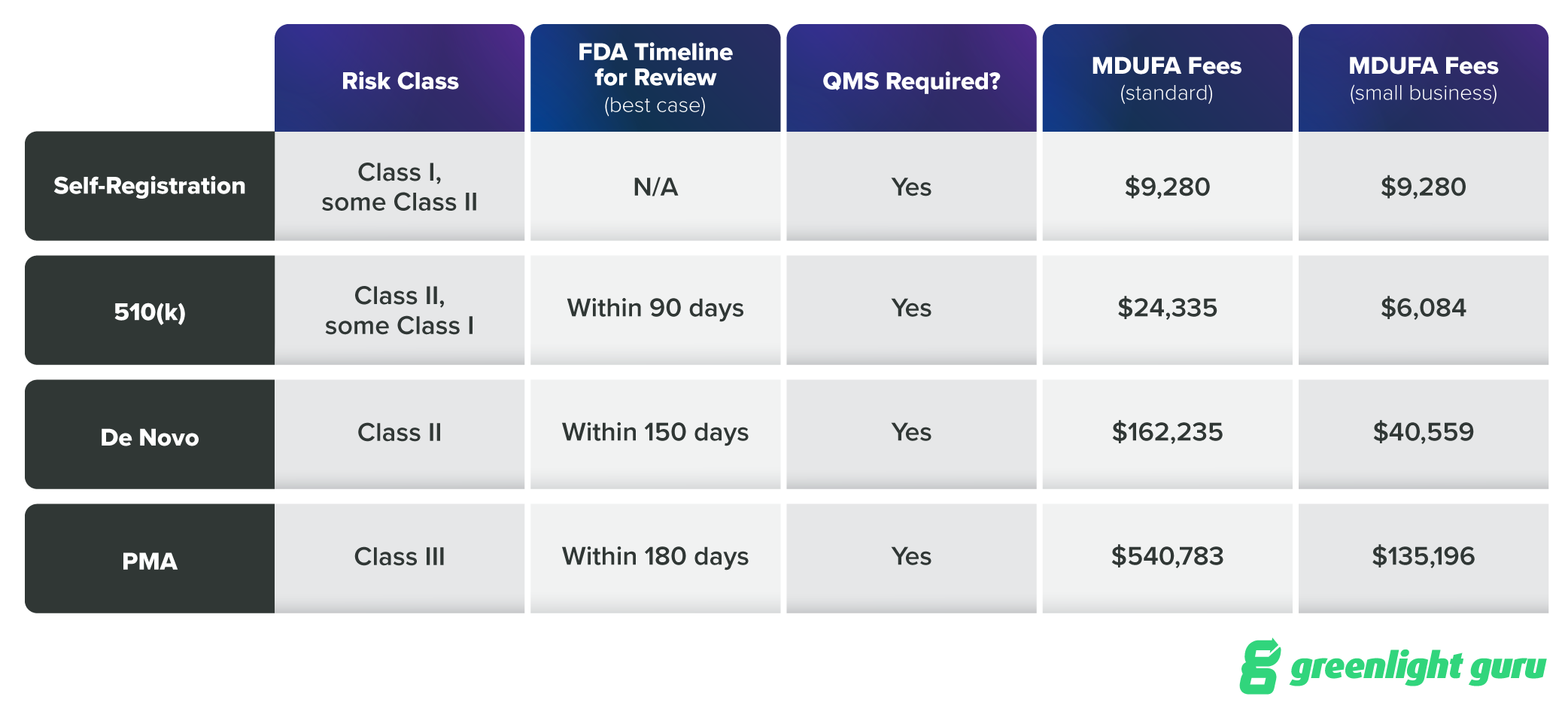
Ideally, you’ll be thinking about your regulatory strategy during design and development, as changes to the device may change its risk, and therefore your regulatory pathway. But your regulatory strategy doesn’t have to be entirely reactive. Choosing to request De Novo classification, for instance, can be a strategic move; we discuss this in length on this episode of the Global Medical Device Podcast.
Whatever route to market you take, just make sure that you’re working on it early and fully understand the implications and consequences of the route you take.
Consider your clinical strategy, if necessary
For class I and most class II devices, a clinical trial is not necessary to get FDA clearance. For class III devices, however, you will need to have clinical data to support your PMA submission.
One thing to remember is that clinical trials always take longer than you think they will. Whether it’s getting IRB approval or recruiting study participants, there are typically delays in the timeline. I’d urge you to plan ahead and build in time for the inevitable delays in carrying out your study.
Something else to consider is that while the 510(k) pathway doesn’t typically require a clinical trial, it may be a good idea to plan one anyway. That’s because market access and market adoption are not the same thing. Getting your 510(k) clearance is great, but you’ll need clinical evidence if you want clinicians to begin choosing your device over its predicate.
Communicate with FDA early and often
This point is especially important for moderate to high risk devices that are taking the more rigorous regulatory pathways. The FDA’s Q-Submission Program is available for manufacturers that are taking any of the regulatory pathways to approval that we’ve discussed so far.
There are a number of different formal meeting requests covered by the Q-Submission program, but a Pre-Submission (Pre-Sub) request is one of the most common. A Pre-Sub is a formal written request for feedback from FDA prior to your pre-market submission that you can use to request information on a variety of topics related to your submission.
As FDA states in their guidance document on the Q-Submission program, “Early interaction with FDA on planned non-clinical and clinical studies and careful consideration of FDA’s feedback may improve the quality of subsequent submissions, shorten total review times, and facilitate the development process for new devices.”
While this is not meant to be a general pre-review of your submission, it is there to help you get answers to questions directly from FDA—which will contribute to a smooth submission and, hopefully, approval.
BONUS RESOURCE: Click here to download a free 510(k) submission checklist!
One big factor in the medical device approval process? Having the right QMS
No matter what regulatory pathway you choose, there are some things that simply won’t change—like, for example, the need for a Quality Management System. Whatever regulatory pathway you take to market approval, your QMS needs to be fully traceable and ready for audits or inspections at all times.
That’s why at Greenlight Guru, we offer the leading QMS software designed by medical device professionals specifically for medical device professionals. Our comprehensive, out-of-the-box solution is based on the latest FDA and ISO standards, as well as best practices of medical device manufacturers who lead the industry in production of high-quality devices.
When you choose Greenlight Guru, you’re choosing the QMS platform that over 1,000 medical device companies trust to keep them compliant and audit-ready. So, if you’re ready to see what a purpose-built QMS can do for your business, then get your free demo of Greenlight Guru today.
BONUS RESOURCE:
FDA 510(k) Submission Checklist
FDA 510(k) Submission Checklist






