When was your last FDA inspection?You may believe that the likelihood of an FDA inspection happening to your company is low.
However, medical device companies registered with FDA with class II and class III products are subject to mandatory inspections at least once in every two year period.
FDA Medical Device QS Surveillance Inspections CY2008 – CY2015

(Source: 2015 Annual FDA Medical Device Quality System Data)
Yes, I know that not every med device firm gets inspected every two years.
If FDA showed up today, would you be ready? Either way, I recommend you do a little bit of homework and spend some time to prepare for an FDA inspection.
(Interesting to note that the number of FDA inspections for foreign / outside U.S. firms is very much on the rise and an initiative of the FDA.)
Even if you are still in development with your very first device waiting on the elusive 510(k) clearance, you should still do your homework and prepare for an FDA inspection.

In fact, now is the best time to establish your QMS and ensure processes are in place and being followed.
Here’s why.
Having an FDA inspection should not ever be a surprise. This is part of what you should expect for being in the medical device industry.
The scope of the FDA inspection also should not ever be a surprise. FDA quality system regulations are published and have been in place for a long time (refer to FDA 21 CFR part 820).
Additionally, FDA tells you how they will inspect you through their, “Guide to Inspections of Quality Systems” (also know as QSIT).
Further, year after year, FDA publishes data on results of FDA inspections and highlights the areas where there are the biggest issues across the entire industry.
Let me share some quick facts from the FDA regarding 2015 medical device inspections
-
FDA conducted 2,104 medical device inspections
-
1484 domestic
-
620 foreign
-
FDA issued Form 483s to 924 medical device companies
-
A total of 3,525 483 observations were cited for 21 CFR part 820 quality system regulations
-
FDA issued Warning Letters to 121 medical device companies (interestingly about 50% were foreign establishments)
-
A total of 690 warning letter citations were issued regarding 21 CFR part 820 deficiencies
FDA Medical Device Inspection QS Subsystems
FDA reports medical device inspection results according to QS subsystems.
Corrective and Preventive Action (CAPA)
“Each manufacturer shall establish and maintain procedures for implementing corrective
and preventive action Each manufacturer shall maintain processes to address non-conforming product and establish and maintain complaint files. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit.”
The related sections of the CFR included in reporting for this subsystem are:
Production and Process Controls (P&PC)
“Each manufacturer is required to develop, conduct, control, and monitor production
processes to ensure that a device conforms to its specifications. Where deviations from device specifications could occur as a result of the manufacturing process, the manufacturer shall establish and maintain process control procedures that describe any process controls necessary to ensure conformance to specifications. In addition to process controls, this subsection includes purchasing controls, labeling, packaging, handling, storage, and installation.”
The related sections of the CFR included in reporting for this subsystem are:
Management Controls (MGMT)
“Management is responsible for establishing policy and objectives for, and commitment to, quality. The QS regulation requires that each manufacturer establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the GMP requirements. To meet these regulatory requirements, manufacturers are required to provide adequate resources, including the assignment of trained personnel for management,
performance of work, and assessment activities, including internal quality audits.”
The related sections of the CFR included in reporting for this subsystem are:
Design Controls (DES)
“Each manufacturer is required by regulation to establish and maintain design control procedures for any class III or class II device, and a selected group of class I devices. The design control procedures ensure that specified design requirements are met.”
The related sections of the CFR included in reporting for this subsystem is 820.30 - Design Controls.
Document Controls (DOC)
“Each manufacturer is required to establish and maintain procedures to control the documents for approval and distribution as well as changes. Manufacturers are also responsible for creating and maintaining the Device Master Record, the Device History Record and the Quality System Record.”
The related sections of the CFR included in reporting for this subsystem are:
Review of 2015 FDA Inspectional Data
Breakdown of FDA 483 Observations
In 2015, FDA issued Form 483 Inspectional Observations to 924 medical device companies, resulting in 3,525 specific 483s relating to 21 CFR part 820.
Let me share the breakdown of FDA 483 observations according to the QS subsystems.
FDA Inspectional Observations CY2004-CY2015 by QS Subsystem

(Source: 2015 Annual FDA Medical Device Quality System Data)
The past 12 years of FDA medical device inspectional data shows a pretty predictable trend, according to the FDA QS subsystem.
Let me dive a bit deeper and breakdown this inspectional data according to CFR clause and highlight the top 9 specific areas that led to 483 observations in 2015.
Top 9 Reasons for 483 Observations in 2015 by CFR

Breakdown of FDA Warning Letters
In 2015, FDA issued 121 warning letters to medical device companies, resulting in 690 citations against 21 CFR part 820.
Let me dive a bit deeper and breakdown the 2015 warning letter citations according to CFR clause and highlight the top 9 specific areas.
Top 9 Reasons for Warning Letter Citations in 2015 by CFR Clause

Interestingly, 820.50 - Purchasing Controls is one of the top 4 areas of warning letter citations yet only represents < 1% of 483 observations.
Overall Problem Areas for Medical Device Companies
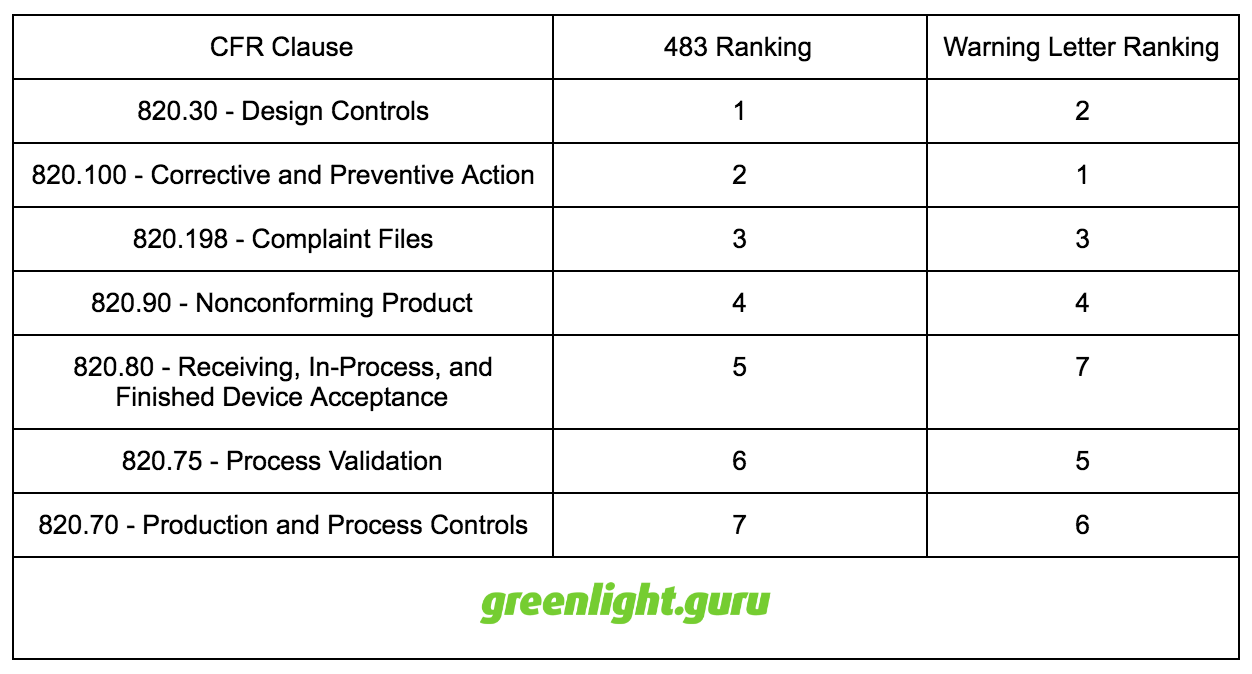
As the data from 2015 FDA 483 observations and warning letters indicates, there are a several key areas for medical device firms to address.
The table below shows the top 7 problem areas according to the specific 21 CFR 820 clause.
Top 7 Problem Areas by CFR Clause